Translational Neurodegeneration Section "Albrecht Kossel", Department of Neurology, University Medical Center Rostock, 18147 Rostock, Germany.
Department of Neurology, University Hospital Carl Gustav Carus, Technische Universität Dresden, 01307 Dresden, Germany.
Int J Mol Sci. 2021 Nov 10;22(22):12184. doi: 10.3390/ijms222212184.
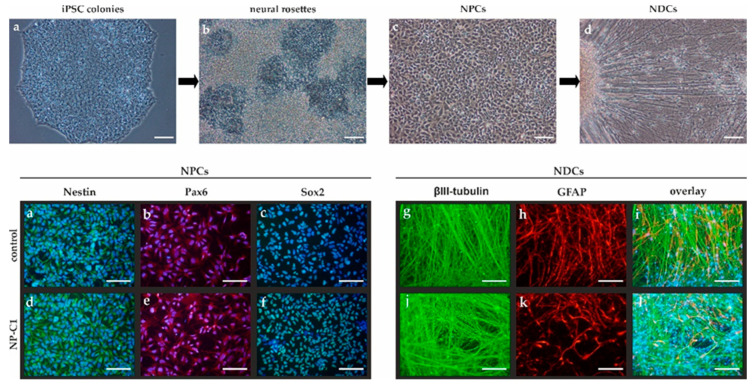
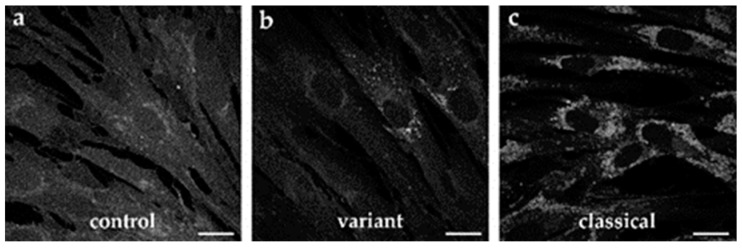
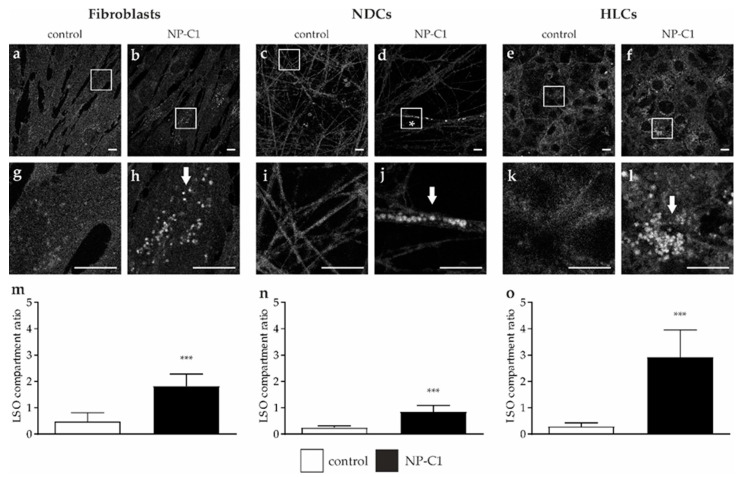
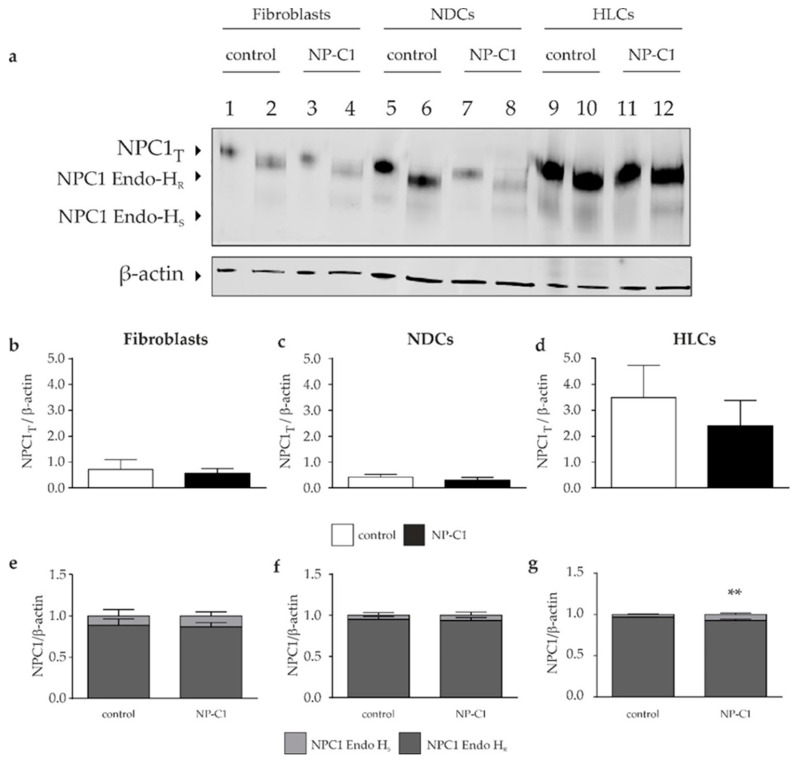
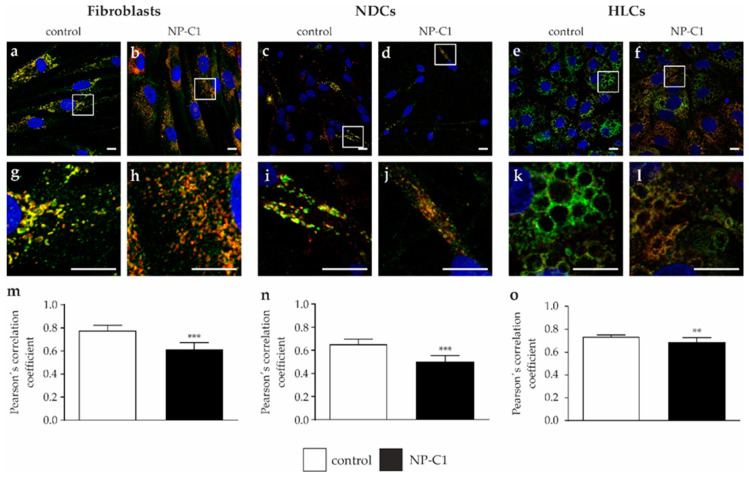
Niemann-Pick disease type C1 (NP-C1) is a rare lysosomal storage disorder caused by autosomal recessive mutations in the gene. Patients display a wide spectrum on the clinical as well as on the molecular level, wherein a so-called "variant" biochemical phenotype can be observed. Here, we report an in vitro analysis of fibroblasts obtained from an NP-C1 patient carrying the undescribed compound heterozygous mutation p.V1023Sfs15/p.G992R. Since NP-C1 is a neurovisceral disease and the patient suffers from severe neurological as well as hepatic symptoms, we extended our study to neural differentiated and hepatocyte-like cells derived from patient-specific induced pluripotent stem cells. We detected slightly increased intracellular cholesterol levels compared to the control cell line in fibroblasts, neural differentiated and hepatocyte-like cells, suggesting a "variant" biochemical phenotype. Furthermore, the total NPC1 protein, as well as post-ER glycoforms of the NPC1 protein, tended to be reduced. In addition, colocalization analysis revealed a mild reduction of the NPC1 protein in the lysosomes. The patient was diagnosed with NP-C1 at the age of 34 years, after an initial misdiagnosis of schizophrenia. After years of mild and unspecific symptoms, such as difficulties in coordination and concentration, symptoms progressed and the patient finally presented with ataxia, dysarthria, dysphagia, vertical supranuclear gaze palsy, and hepatosplenomegaly. Genetic testing finally pointed towards an NP-C1 diagnosis, revealing the so-far undescribed compound heterozygous mutation p.V1023Sfs15/p.G992R in the gene. In light of these findings, this case provides support for the p.G992R mutation being causative for a "variant" biochemical phenotype leading to an adult-onset type of NP-C1 disease.
尼曼-匹克病 C1 型(NP-C1)是一种罕见的溶酶体贮积病,由 基因的常染色体隐性突变引起。患者在临床和分子水平上表现出广泛的表型,其中可以观察到所谓的“变异”生化表型。在这里,我们报告了对来自携带未描述的复合杂合突变 p.V1023Sfs15/p.G992R 的 NP-C1 患者的成纤维细胞进行的体外分析。由于 NP-C1 是一种神经内脏疾病,患者患有严重的神经和肝脏症状,因此我们将研究扩展到源自患者特异性诱导多能干细胞的神经分化和肝细胞样细胞。与对照细胞系相比,我们在成纤维细胞、神经分化和肝细胞样细胞中检测到细胞内胆固醇水平略有升高,这表明存在“变异”生化表型。此外,NPC1 蛋白的总蛋白以及 NPC1 蛋白的内质网糖基化形式趋于减少。此外,共定位分析显示溶酶体中 NPC1 蛋白的表达略有减少。该患者在 34 岁时被诊断为 NP-C1,此前曾被误诊为精神分裂症。在经历了数年的轻度和非特异性症状,如协调和注意力困难后,症状逐渐进展,患者最终出现共济失调、构音障碍、吞咽困难、垂直核上性眼球运动麻痹和肝脾肿大。基因检测最终指向 NP-C1 诊断,揭示了 基因中迄今为止未描述的复合杂合突变 p.V1023Sfs15/p.G992R。鉴于这些发现,该病例为 p.G992R 突变导致“变异”生化表型从而导致成人发病型 NP-C1 疾病提供了支持。