Xu Chengxian, Yang Chenxi, Ye Qing, Xu Jie, Tong Lingxiao, Zhang Yuchen, Shen Huijun, Lu Zhihong, Wang Jingjing, Lai Enyin, Mao Jianhua, Jiang Pingping
Department of Nephrology, The Children's Hospital, Zhejiang University School of Medicine and National Clinical Research Center for Child Health, Hangzhou, China.
Institute of Genetics and Department of Human Genetics, Zhejiang University School of Medicine, Hangzhou, China.
Front Med (Lausanne). 2021 Dec 16;8:743150. doi: 10.3389/fmed.2021.743150. eCollection 2021.
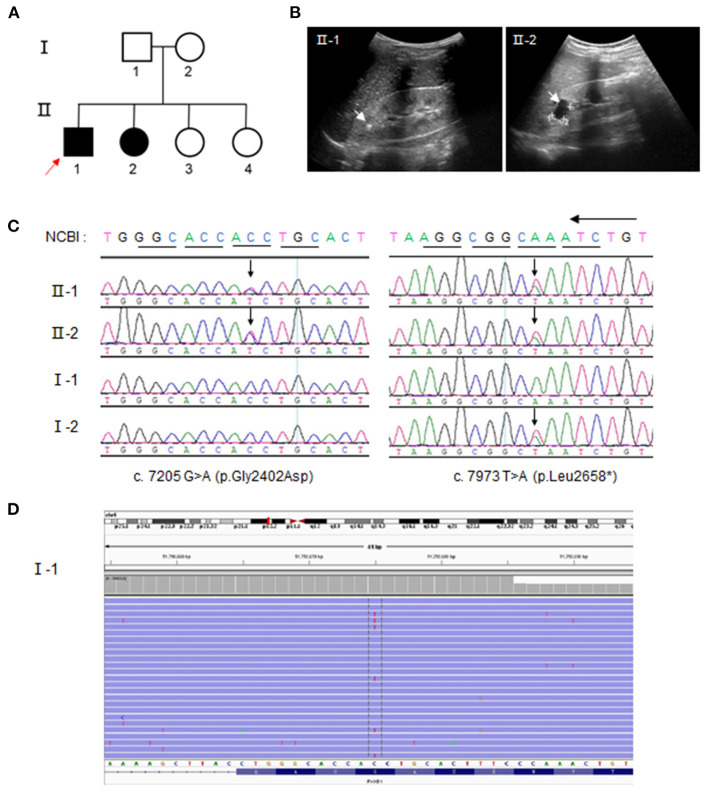
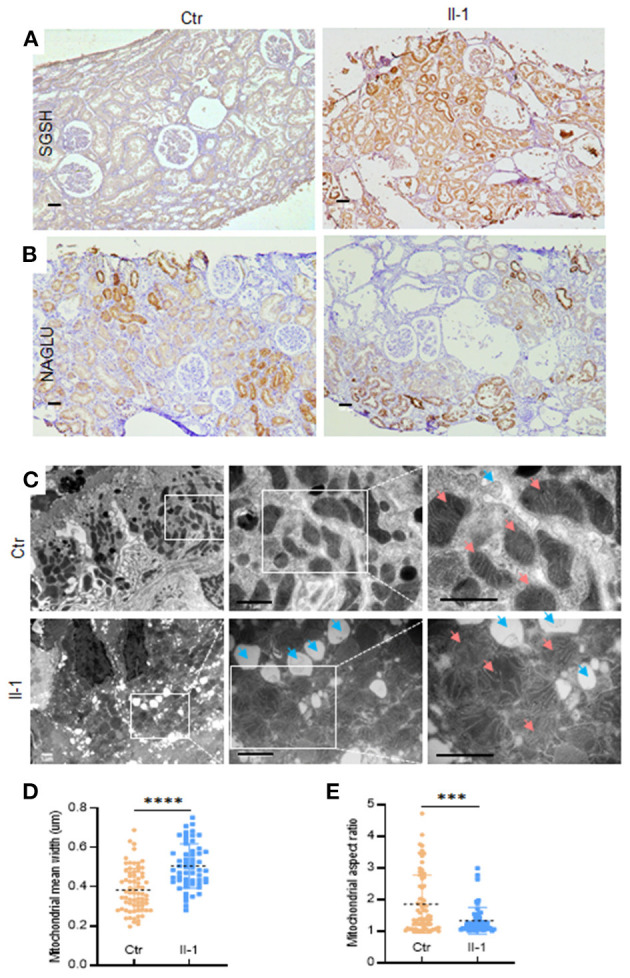
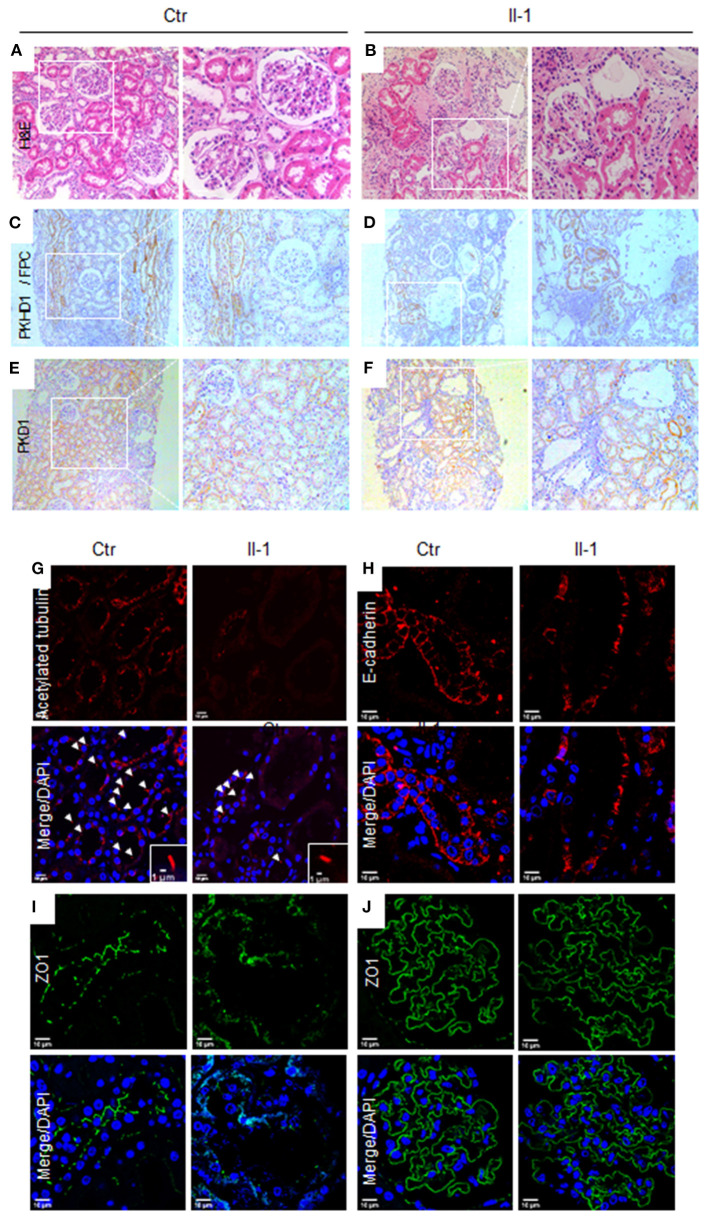
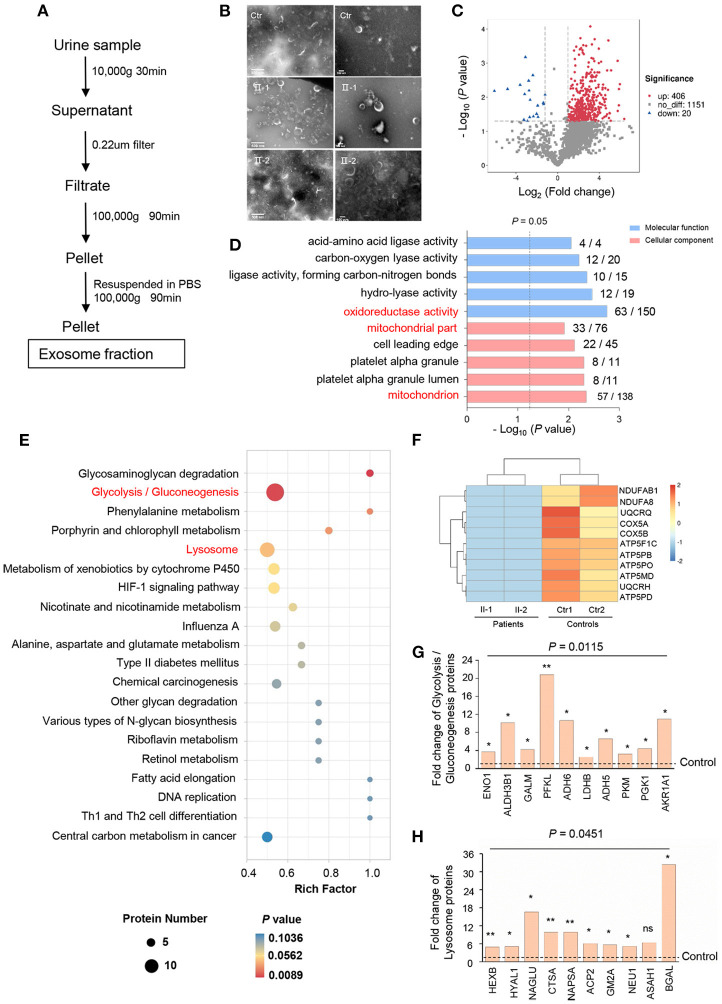
Autosomal recessive polycystic kidney disease (ARPKD) is a severe renal cystic disease caused mainly by the polycystic kidney and hepatic disease 1 (. However, the genetic cause, pathologic features, and mechanism of action of ARPKD are not well known. Here, we identified a family with ARPKD. Two siblings harbored biallelic variants in (c.7205G>A, c.7973T>A). We determined that the "" variant, c.7205G>A, arose from the mosaicism of the father and had a 7.4% level. Pathologic characterization, using biopsy analysis, was evidenced with predominant cystic dilation in proximal tubules, slight ectasia of collecting ducts, defective ciliogenesis, and impaired cell-cell junctions in renal tubules and collecting ducts. Exosome proteomics in the urine from patients with ARPKD were markedly different from those of controls, with the most significant alterations occurring in mitochondrial and lysosomal proteins. Expression of the proteins of OXPHOS was downregulated sharply, in parallel with upregulated expression of the proteins involved in glycolysis in patients with ARPKD. Several lysosomal proteins associated with renal lesions were more abundant in the exosome of the patient than in controls. Moreover, the lysosomal enzyme sulfamidase, which is produced by the gene, was abrupt uniquely in the exosome of the patient. Consistently, swollen mitochondria and abundant lysosomes were visualized in the mutant tubular epithelial cells of patients with mutant . Collectively, these findings provide new insights on the pathophysiology of the polycystic kidney due to PKHD1 deficiency. mosaicism should be considered in genetic testing of ARPKD patients.
常染色体隐性多囊肾病(ARPKD)是一种严重的肾囊性疾病,主要由多囊肾和肝病1基因(PKHD1)引起。然而,ARPKD的遗传病因、病理特征及作用机制尚不清楚。在此,我们鉴定了一个患有ARPKD的家系。两名同胞在PKHD1基因中存在双等位基因变异(c.7205G>A,c.7973T>A)。我们确定,“c.7205G>A”变异源自父亲的嵌合体,其水平为7.4%。通过活检分析进行的病理特征显示,近端小管主要为囊性扩张,集合管轻度扩张,纤毛发生缺陷,肾小管和集合管中的细胞间连接受损。ARPKD患者尿液中的外泌体蛋白质组学与对照组明显不同,最显著的变化发生在线粒体和溶酶体蛋白中。ARPKD患者中氧化磷酸化(OXPHOS)蛋白的表达急剧下调,同时糖酵解相关蛋白的表达上调。与肾损伤相关的几种溶酶体蛋白在患者的外泌体中比对照组更丰富。此外,由SUMF1基因产生的溶酶体酶硫酸酯酶在患者的外泌体中独特地突然出现。一致地,在携带突变PKHD1的患者的突变肾小管上皮细胞中可见肿胀的线粒体和丰富的溶酶体。总体而言,这些发现为PKHD1缺乏导致的多囊肾的病理生理学提供了新的见解。在ARPKD患者的基因检测中应考虑嵌合体情况。