Institute for Pathology, Medical Faculty and University Hospital of Cologne, University of Cologne, Cologne, Germany; Center for Molecular Medicine Cologne (CMMC), University of Cologne, Cologne, Germany.
Institute for Pathology, Medical Faculty and University Hospital of Cologne, University of Cologne, Cologne, Germany.
Cell Mol Gastroenterol Hepatol. 2022;13(6):1701-1716. doi: 10.1016/j.jcmgh.2022.02.013. Epub 2022 Feb 24.
BACKGROUND & AIMS: Liver fibrosis arises from long-term chronic liver injury, accompanied by an accelerated wound healing response with interstitial accumulation of extracellular matrix (ECM). Activated hepatic stellate cells (HSC) are the main source for ECM production. MicroRNA29a (miR-29a) is a crucial antifibrotic miRNA that is repressed during fibrosis, resulting in up-regulation of collagen synthesis.
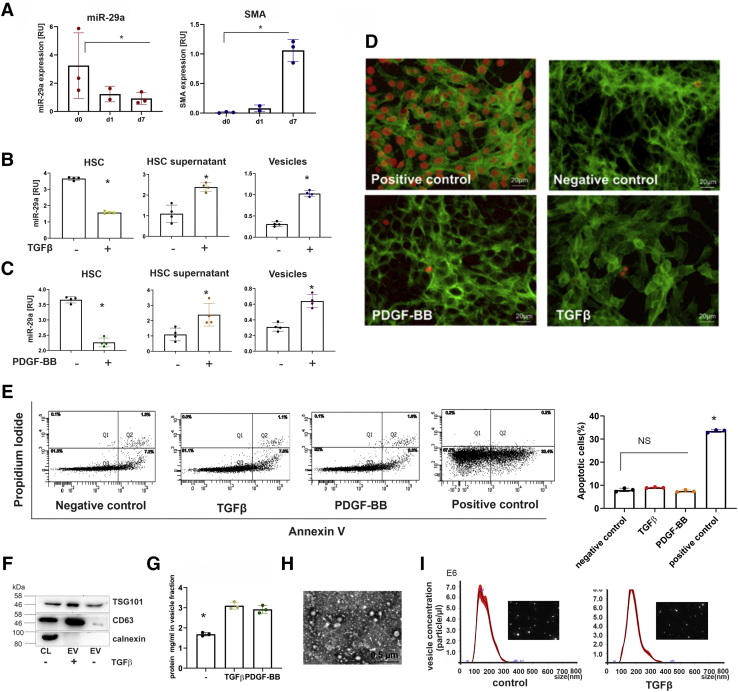
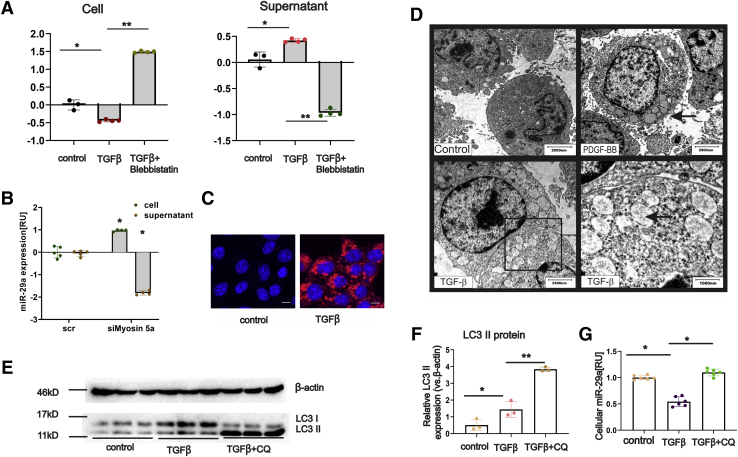
Intracellular and extracellular miRNA levels of primary and immortalized myofibroblastic HSC in response to profibrogenic stimulation by transforming growth factor β (TGFβ) or platelet-derived growth factor-BB (PDGF-BB) or upon inhibition of vesicular transport and autophagy processes were determined by quantitative polymerase chain reaction. Autophagy flux was studied by electron microscopy, flow cytometry, immunoblotting, and immunocytochemistry. Hepatic and serum miR-29a levels were quantified by using both liver tissue and serum samples from a cohort of chronic hepatitis C virus patients and a murine CCl induced liver fibrosis model.
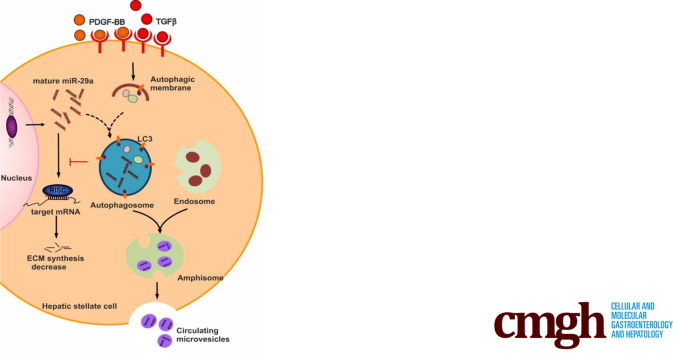
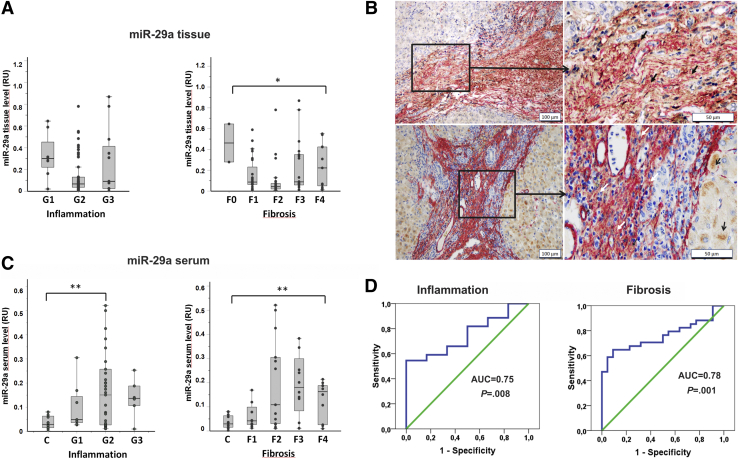
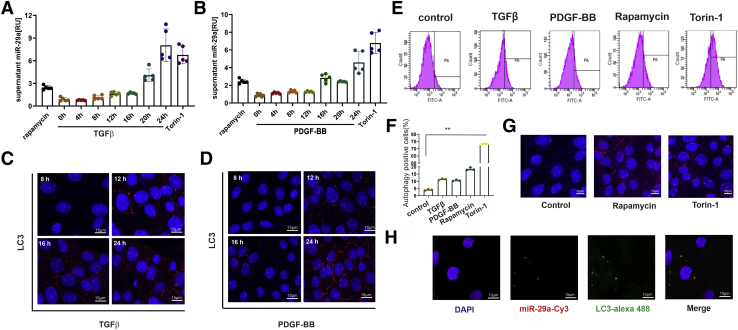
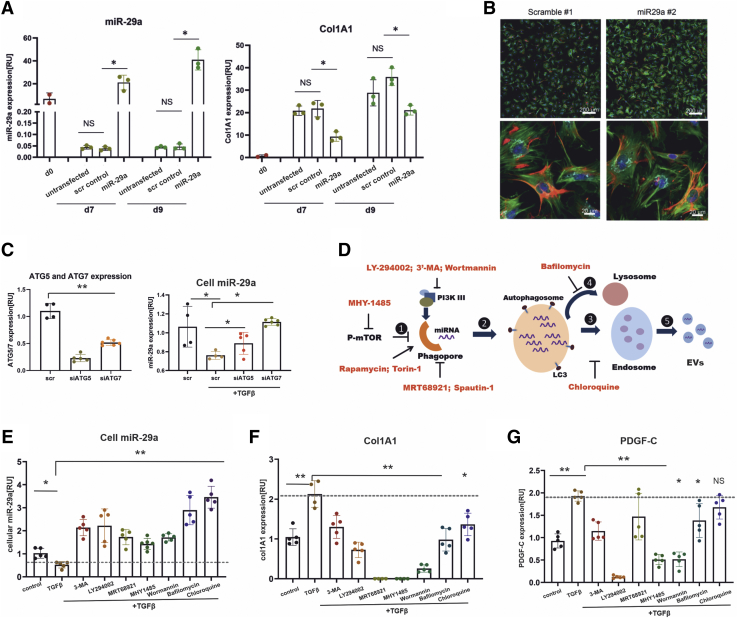
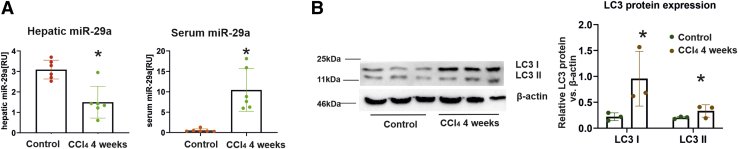
In our study, we show that TGFβ and PDGF-BB resulted in decrease of intracellular miR-29a and a pronounced increase of vesicular miR-29a release into the supernatant. Strikingly, miR-29a vesicular release was accompanied by enhanced autophagic activity and up-regulation of the autophagy marker protein LC3. Moreover, autophagy inhibition strongly prevented miR-29a secretion and repressed its targets' expression such as Col1A1. Consistently, hepatic miR-29a loss and increased LC3 expression in myofibroblastic HSC were associated with increased serum miR-29a levels in CCl-treated murine liver fibrosis and specimens of hepatitis C virus patients with chronic liver disease.
We provide evidence that activation-associated autophagy in HSC induces release of miR-29a, whereas inhibition of autophagy represses fibrogenic gene expression in part through attenuated miR-29a secretion.
肝纤维化由长期慢性肝损伤引起,伴有细胞外基质(ECM)间质积累的加速伤口愈合反应。活化的肝星状细胞(HSC)是 ECM 产生的主要来源。微小 RNA29a(miR-29a)是一种重要的抗纤维化 miRNA,在纤维化过程中受到抑制,导致胶原合成上调。
通过定量聚合酶链反应测定转化生长因子β(TGFβ)或血小板衍生生长因子-BB(PDGF-BB)致纤维化刺激或抑制囊泡运输和自噬过程对原代和永生化肌成纤维细胞 HSC 的细胞内和细胞外 miRNA 水平。通过电子显微镜、流式细胞术、免疫印迹和免疫细胞化学研究自噬流。通过使用慢性丙型肝炎病毒患者的肝组织和血清样本以及 CCl 诱导的肝纤维化模型,定量测定肝和血清 miR-29a 水平。
在我们的研究中,我们表明 TGFβ 和 PDGF-BB 导致细胞内 miR-29a 减少,并显著增加囊泡 miR-29a 释放到上清液中。引人注目的是,miR-29a 囊泡释放伴随着增强的自噬活性和自噬标志物蛋白 LC3 的上调。此外,自噬抑制强烈阻止 miR-29a 分泌并抑制其靶基因如 Col1A1 的表达。一致地,在 CCl 处理的肝纤维化小鼠模型和丙型肝炎病毒患者慢性肝病的标本中,肌成纤维细胞 HSC 中与激活相关的自噬导致 miR-29a 丢失和 LC3 表达增加,与血清 miR-29a 水平升高相关。
我们提供的证据表明,HSC 中激活相关的自噬诱导 miR-29a 的释放,而自噬抑制通过减弱 miR-29a 的分泌部分抑制成纤维基因表达。