Wang Jing-Yang, Tang Bin, Sheng Wen-Xiang, Hua Li-Dong, Zeng Yang, Fan Cui-Xia, Deng Wei-Yi, Gao Mei-Mei, Zhu Wei-Wen, He Na, Su Tao
Institute of Neuroscience, The Second Affiliated Hospital of Guangzhou Medical University, Guangzhou, China.
Key Laboratory of Neurogenetics and Channelopathies, Ministry of Education of China, Guangzhou, China.
Front Mol Neurosci. 2022 Mar 14;15:828846. doi: 10.3389/fnmol.2022.828846. eCollection 2022.
Naturally occurring in-frame deletion is a unique type of genetic variations, causing the loss of one or more amino acids of proteins. A number of in-frame deletion variants in an epilepsy-associated gene , encoding voltage gated sodium channel alpha unit 1.1 (Na1.1), have been reported in public database. In contrast to the missense and truncation variants, the in-frame deletions in remains largely uncharacterized.
We summarized the basic information of forty-four in-frame deletion variants and performed further analysis on six variants identified in our cases with epilepsy. Mutants of the six in-frame deletions and one truncating variant used as comparison were generated and co-transfected with beta-1 and -2 subunits in tsA201 cells, followed by patch clamp recordings.
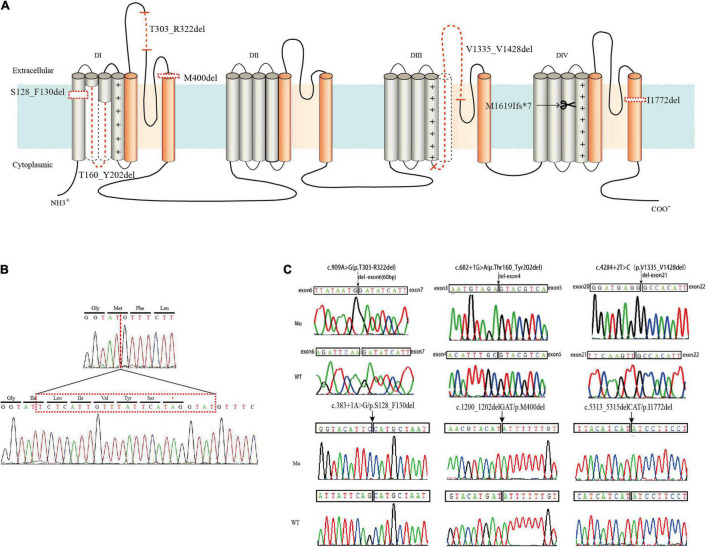
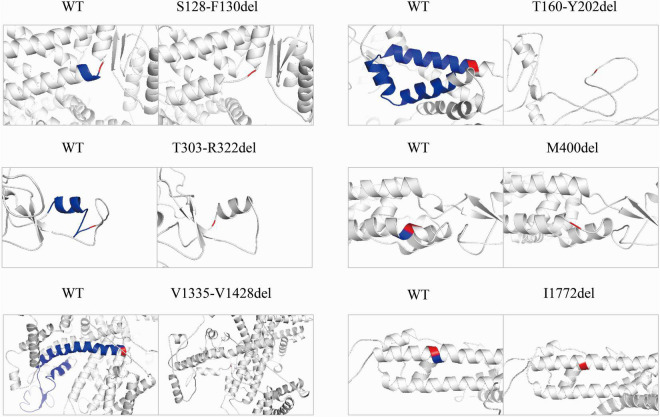
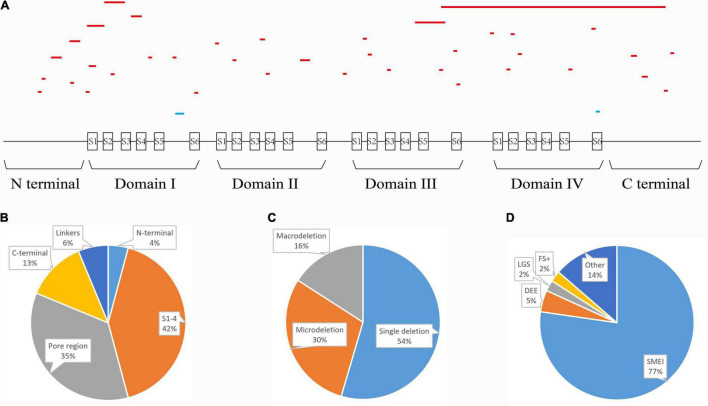
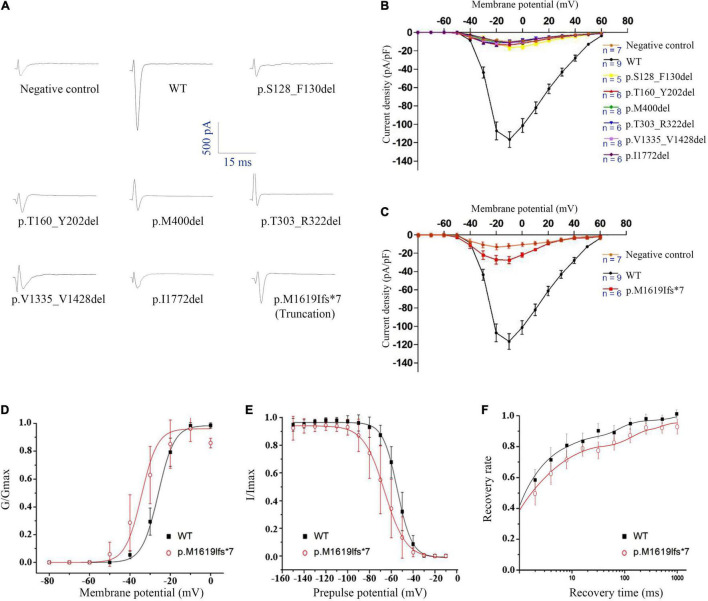
Reviewing all the in-frame deletions showed that they spread over the entire Na1.1 protein, without obvious "hot spots." The dominant type (54%) was single residue loss. There was no obvious relationship between the length or locations of deletions and their clinical phenotypes. The six in-frame deletions were two single residue deletions (p.M400del and p.I1772del), one microdeletion (p.S128_F130del) and three macrodeletions (p.T303_R322del, p.T160_Y202del, and p.V1335_V1428del). They scatter and affect different functional domains, including transmembrane helices, pore region, and P-loop. Electrophysiological recordings revealed no measurable sodium current in all of the six mutants. In contrast, the truncating mutant p.M1619Ifs*7 that loses a long stretch of peptides retains partial function.
The complete loss-of-function in these shortened, abnormal mutants indicates that Na1.1 protein is a highly accurate structure, and many of the residues have no redundancy to ion conductance. In-frame deletions caused particularly deleterious effect on protein function possibly due to the disruption of ordered residues.
自然发生的框内缺失是一种独特的基因变异类型,会导致蛋白质中一个或多个氨基酸的丢失。在公共数据库中已报道了癫痫相关基因(编码电压门控钠通道α亚基1.1,即Na1.1)中的一些框内缺失变异。与错义变异和截短变异不同,Na1.1中的框内缺失在很大程度上仍未得到充分研究。
我们总结了44个框内缺失变异的基本信息,并对在我们的癫痫病例中鉴定出的6个变异进行了进一步分析。构建了6个框内缺失突变体和1个用作对照的截短变异体,并在tsA201细胞中与β-1和β-2亚基共转染,随后进行膜片钳记录。
回顾所有框内缺失发现,它们分布在整个Na1.1蛋白上,没有明显的“热点”。主要类型(54%)是单个残基缺失。缺失的长度或位置与其临床表型之间没有明显关系。这6个框内缺失分别是两个单个残基缺失(p.M400del和p.I1772del)、一个微缺失(p.S128_F130del)和三个大缺失(p.T303_R322del、p.T160_Y202del和p.V1335_V1428del)。它们分散并影响不同的功能结构域,包括跨膜螺旋、孔区和P环。电生理记录显示,所有6个突变体均未检测到可测量的钠电流。相比之下,丢失一长段肽段的截短突变体p.M1619Ifs*7保留了部分功能。
这些缩短的异常突变体中功能的完全丧失表明,Na1.1蛋白是一种高度精确的结构,许多残基对离子传导没有冗余。框内缺失可能由于有序残基的破坏而对蛋白质功能造成特别有害的影响。