Sanchez-Villalobos Maria, Blanquer Miguel, Moraleda Jose M, Salido Eduardo J, Perez-Oliva Ana B
Hematology Service, Virgen de la Arrixaca University Hospital, Murcia, Spain.
Biomedical Research Institute of Murcia (IMIB), Murcia, Spain.
Front Med (Lausanne). 2022 Apr 12;9:880752. doi: 10.3389/fmed.2022.880752. eCollection 2022.
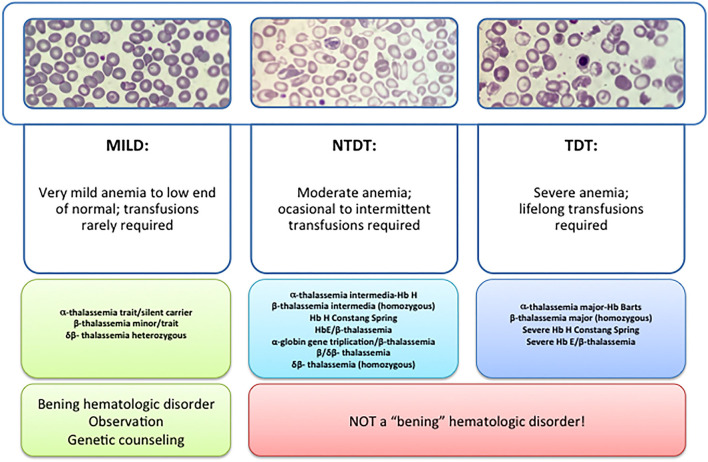
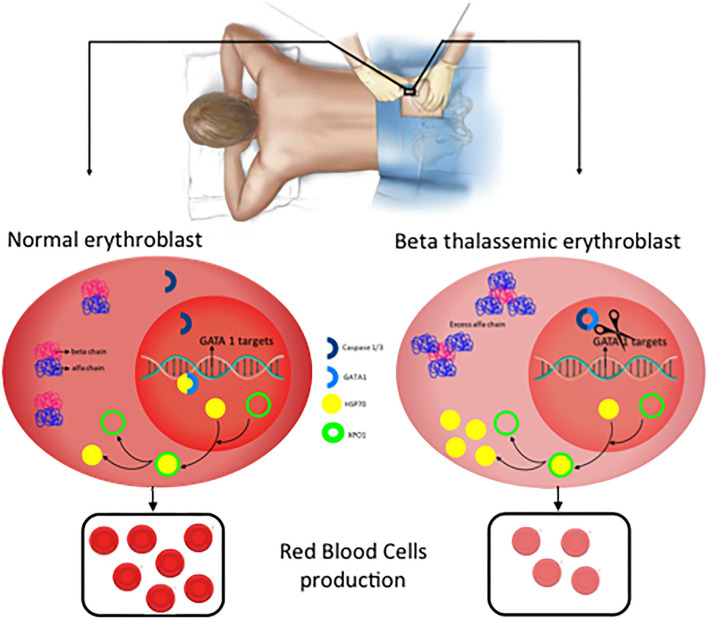
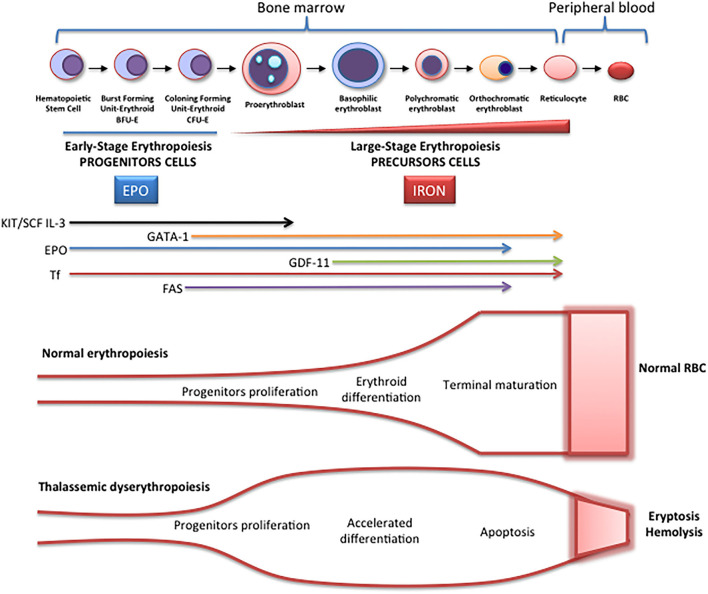
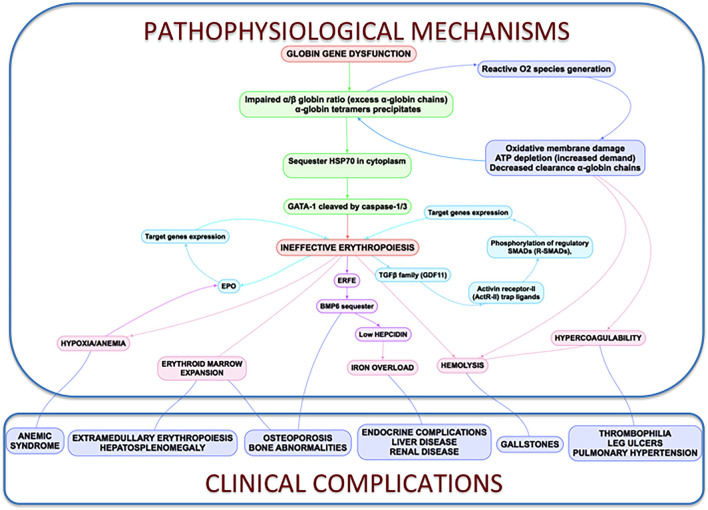
β-thalassemia is a disease caused by genetic mutations including a nucleotide change, small insertions or deletions in the β-globin gene, or in rare cases, gross deletions into the β-globin gene. These mutations affect globin-chain subunits within the hemoglobin tetramer what induces an imbalance in the α/β-globin chain ratio, with an excess of free α-globin chains that triggers the most important pathogenic events of the disease: ineffective erythropoiesis, chronic anemia/chronic hypoxia, compensatory hemopoietic expansion and iron overload. Based on advances in our knowledge of the pathophysiology of β-thalassemia, in recent years, emerging therapies and clinical trials are being conducted and are classified into three major categories based on the different approach features of the underlying pathophysiology: correction of the α/β-globin disregulation; improving iron overload and reverse ineffective erythropoiesis. However, pathways such as the dysregulation of transcriptional factors, activation of the inflammasome, or approach to mechanisms of bone mineral loss, remain unexplored for future therapeutic targets. In this review, we update the main pathophysiological pathways involved in β-thalassemia, focusing on the development of new therapies directed at new therapeutic targets.
β地中海贫血是一种由基因突变引起的疾病,这些突变包括β珠蛋白基因中的核苷酸变化、小的插入或缺失,或在罕见情况下,β珠蛋白基因的大片段缺失。这些突变影响血红蛋白四聚体内的珠蛋白链亚基,导致α/β珠蛋白链比例失衡,游离α珠蛋白链过量,从而引发该疾病最重要的致病事件:无效造血、慢性贫血/慢性缺氧、代偿性造血扩张和铁过载。基于我们对β地中海贫血病理生理学认识的进展,近年来,正在开展新出现的治疗方法和临床试验,并根据潜在病理生理学的不同方法特点分为三大类:纠正α/β珠蛋白失调;改善铁过载和逆转无效造血。然而,转录因子失调、炎性小体激活或骨矿物质丢失机制等途径,仍是有待探索的未来治疗靶点。在本综述中,我们更新了β地中海贫血所涉及的主要病理生理途径,重点关注针对新治疗靶点的新疗法的开发。