Lim Chelsey Chaehee, Vockley Jerry, Ujah Otobo, Kirby Russell S, Edick Mathew J, Berry Susan A, Arnold Georgianne L
University of Pittsburgh School of Medicine, USA.
UPMC Children's Hospital of Pittsburgh, USA.
Mol Genet Metab Rep. 2022 Jun 3;32:100884. doi: 10.1016/j.ymgmr.2022.100884. eCollection 2022 Sep.
Mitochondrial trifunctional protein deficiency (TFPD) and isolated long chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD) are two related defects of fatty acid β -oxidation. While NBS has decreased mortality, morbidity remains significant. Additionally, the relationship of genotype to clinical outcome remains unclear. To better understand these issues, we collected natural history data for these conditions by reviewing seven years of retrospective data from 45 cases of TFPD or LCHADD in the Inborn Errors of Metabolism - Information System.
Available data included age at database entry, last datapoint, and development of various complications. Data were analyzed by clinical assigned diagnosis (LCHADD or TFPD), subdivided by method of ascertainment (newborn screening-NBS, or other than by newborn screening-NNBS), then re-analyzed based on four genotype groups: homozygous c.1528GC (p.E510Q) (common LCHAD variant); heterozygous c.1528GC (p.E510Q), other variants; and variants.
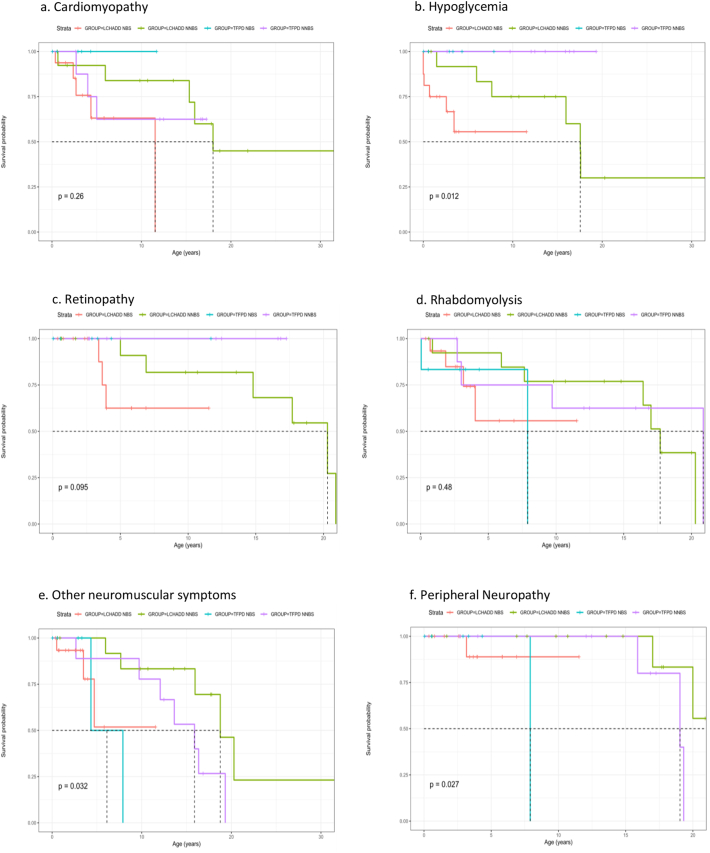
Forty-five patients from birth to 34 years of age were analyzed by assigned diagnosis (30 LCHADD and 15 TFPD) and method of ascertainment. Thirty had further analysis by genotype (22 biallelic variants and 8 biallelic variants). With regards to maternal complications, retinopathy, cardiomyopathy and hypoglycemia, patients with biallelic variants (with or without the common LCHAD variant) manifest a traditional LCHADD phenotype, while those with gene variants more commonly reported neuromusculoskeletal type TFPD phenotype. While retinopathy, rhabdomyolysis and peripheral neuropathy tended to present later in childhood, many features including initial report of cardiomyopathy and hypoglycemia presented across a wide age spectrum.
This study demonstrates the utility of genotypic confirmation of patients identified with LCHADD/TFPD as variants in the and genes lead to different symptom profiles. In our data, biallelic variants conferred a LCHADD phenotype, regardless of the presence of the common LCHAD variant.
线粒体三功能蛋白缺乏症(TFPD)和孤立性长链3-羟酰基辅酶A脱氢酶缺乏症(LCHADD)是脂肪酸β氧化的两种相关缺陷。虽然新生儿筛查(NBS)降低了死亡率,但发病率仍然很高。此外,基因型与临床结果之间的关系仍不清楚。为了更好地理解这些问题,我们通过回顾代谢性遗传病信息系统中45例TFPD或LCHADD患者的七年回顾性数据,收集了这些疾病的自然史数据。
可用数据包括进入数据库时的年龄、最后一个数据点以及各种并发症的发生情况。数据按临床指定诊断(LCHADD或TFPD)进行分析,再按确诊方法(新生儿筛查-NBS或非新生儿筛查-NNBS)细分,然后基于四个基因型组重新分析:纯合子c.1528G>C(p.E510Q)(常见LCHAD变异);杂合子c.1528G>C(p.E510Q)、其他变异;以及变异。
对45例年龄从出生到34岁的患者按指定诊断(30例LCHADD和15例TFPD)和确诊方法进行分析。30例患者进一步按基因型分析(22例双等位基因变异和8例双等位基因变异)。关于母亲并发症、视网膜病变、心肌病和低血糖,双等位基因变异患者(有或无常见LCHAD变异)表现出传统的LCHADD表型,而有基因变异的患者更常报告神经肌肉骨骼型TFPD表型。虽然视网膜病变、横纹肌溶解和周围神经病变往往在儿童期后期出现,但许多特征包括心肌病和低血糖的首次报告在很宽的年龄范围内都有出现。
本研究证明了对确诊为LCHADD/TFPD的患者进行基因型确认的实用性,因为基因和基因中的变异会导致不同的症状谱。在我们的数据中,双等位基因变异赋予了LCHADD表型,无论是否存在常见的LCHAD变异。