Department of Chemistry, University of Rome "La Sapienza", Piazzale Aldo Moro 5, Rome 00185, Italy.
Biomolecular Mass Spectrometry and Proteomics, Bijvoet Center for Biomolecular Research and Utrecht Institute for Pharmaceutical Sciences, Utrecht University, Padualaan 8, Utrecht 3584 CH, The Netherlands.
Anal Chem. 2022 Jul 5;94(26):9234-9241. doi: 10.1021/acs.analchem.1c05621. Epub 2022 Jun 17.
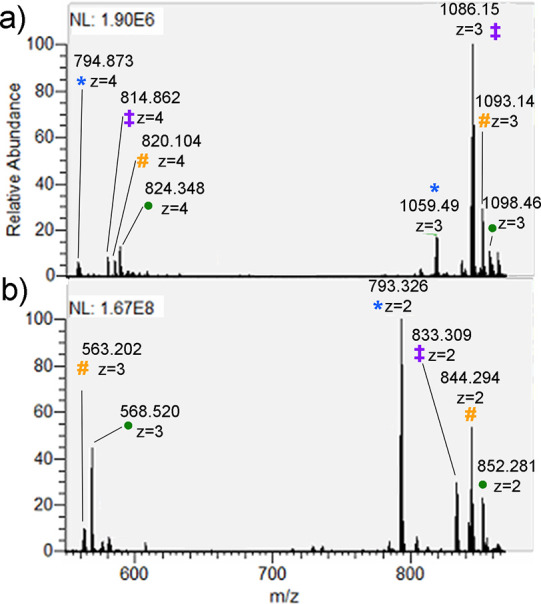
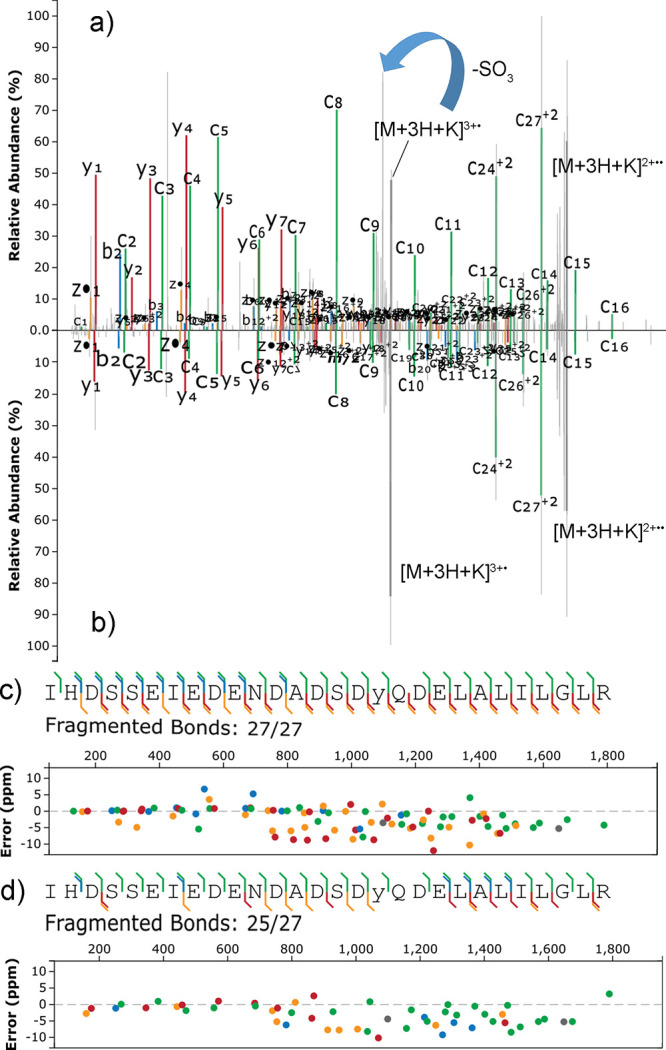
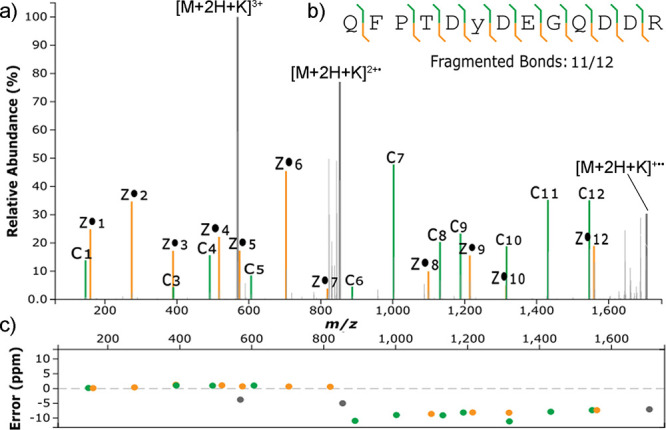
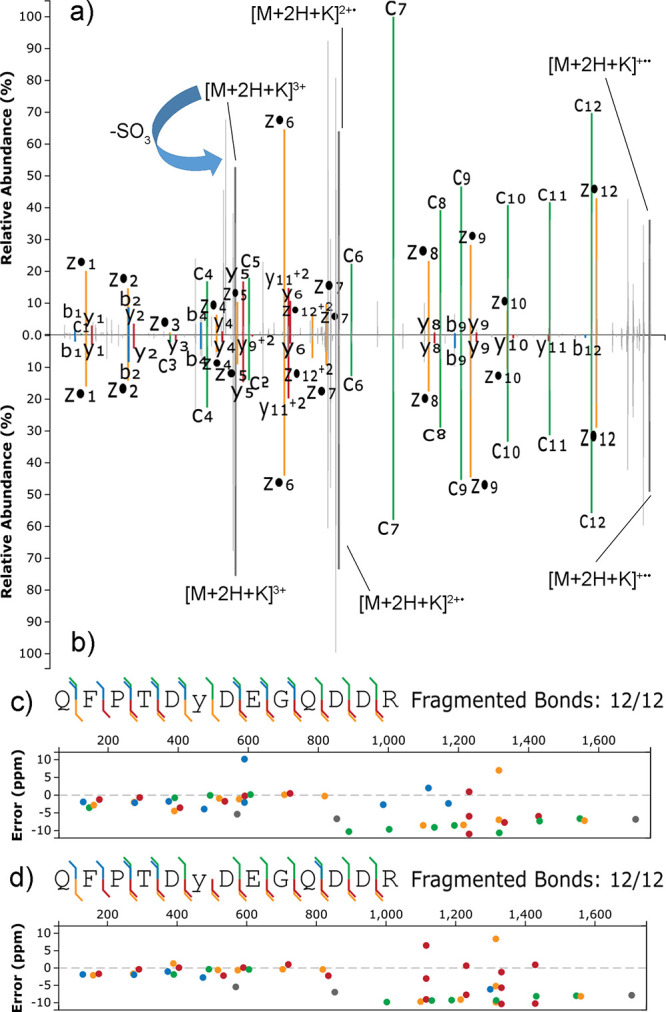
Site localization of protein sulfation by high-throughput proteomics remains challenging despite the technological improvements. In this study, sequence analysis and site localization of sulfation in tryptic peptides were determined under a conventional nano-liquid chromatography-mass spectrometry configuration. Tryptic sulfopeptide standards were used to study different fragmentation strategies, including collision-induced dissociation (CID), higher-energy collisional dissociation (HCD), electron-transfer dissociation (ETD), electron-transfer/higher-energy collision dissociation (EThcD), and electron-transfer/collision-induced dissociation (ETciD), in the positive ionization mode. Sulfopeptides displayed only neutral loss of SO under CID, while the sequence could be determined for all other tested fragmentation techniques. Results were compared to the same sequences with phosphotyrosine, indicating important differences, as the sequence and modification localization could be studied by all fragmentation strategies. However, the use of metal adducts, especially potassium, provided valuable information for sulfopeptide localization in ETD and ETD-hybrid strategies by stabilizing the modification and increasing the charge state of sulfopeptides. In these conditions, both the sequence and localization could be obtained. In-source neutral loss of SO under EThcD provided diagnostic peaks suitable to distinguish the sulfopeptides from the nearly isobaric phosphopeptides. Further confirmation on the modification type was found in the negative ionization mode, where phosphopeptides always had the typical phosphate product ion corresponding to PO.
尽管技术不断进步,但通过高通量蛋白质组学进行蛋白质硫酸化的定位仍然具有挑战性。在这项研究中,我们在常规纳流液相色谱-质谱配置下,确定了硫酸化肽的序列分析和定位。我们使用胰蛋白酶硫酸肽标准品来研究不同的碎片化策略,包括碰撞诱导解离(CID)、高能碰撞解离(HCD)、电子转移解离(ETD)、电子转移/高能碰撞解离(EThcD)和电子转移/碰撞诱导解离(ETciD),均在正离子模式下进行。在 CID 下,硫酸肽仅表现为 SO 的中性丢失,而所有其他测试的碎片化技术都可以确定序列。结果与具有磷酸酪氨酸的相同序列进行了比较,表明存在重要差异,因为所有碎片化策略都可以研究序列和修饰定位。然而,金属加合物的使用,特别是钾,通过稳定修饰和增加硫酸肽的电荷状态,为 ETD 和 ETD-混合策略中的硫酸肽定位提供了有价值的信息。在这些条件下,既可以获得序列,也可以获得定位。在 EThcD 中的源内 SO 中性丢失提供了合适的诊断峰,可将硫酸肽与几乎等摩尔质量的磷酸肽区分开来。在负离子模式下,进一步确认了修饰类型,其中磷酸肽总是具有对应于 PO 的典型磷酸产物离子。