Waheed Waqar, Newman Eric, Aboukhatwa Marwa, Moin Maryam, Tandan Rup
Department of Neurological Sciences, The University of Vermont and the University of Vermont Medical Center, Burlington, VT, USA.
Pharmacotherapy Department, University of Vermont Medical Center, Burlington, VT, USA.
Ther Clin Risk Manag. 2022 Jul 12;18:699-719. doi: 10.2147/TCRM.S266031. eCollection 2022.
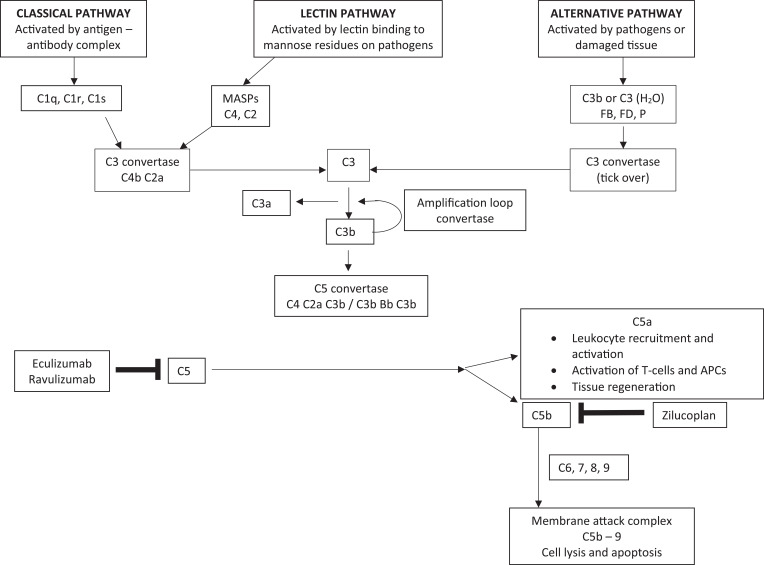
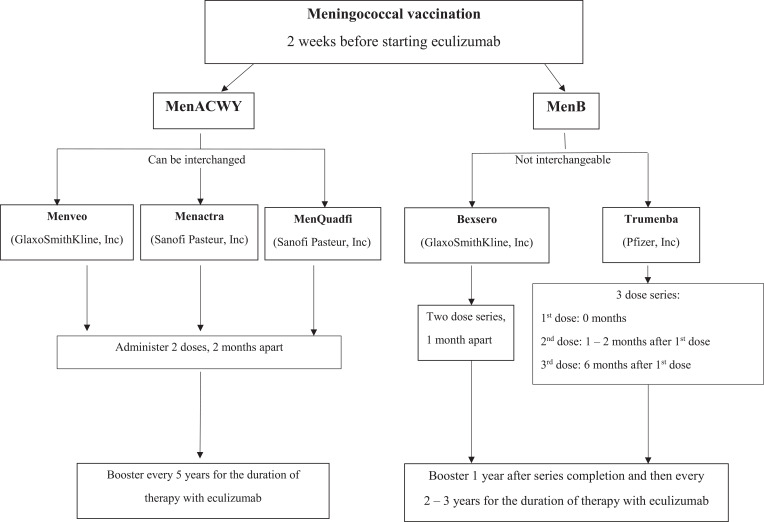
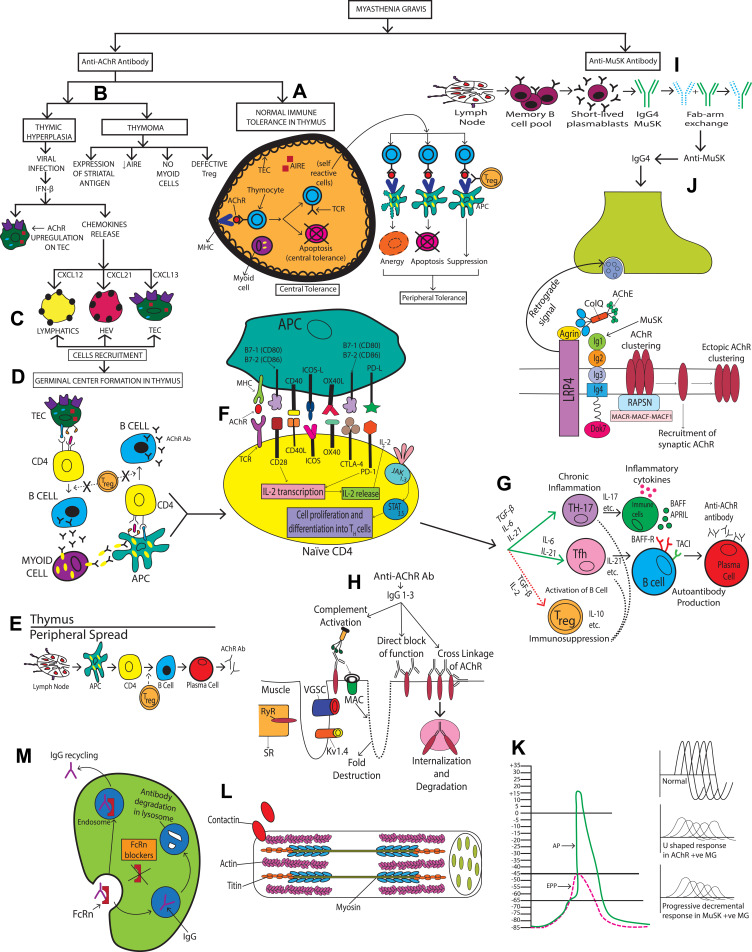
Myasthenia gravis (MG) is a rare autoimmune disorder caused by specific autoantibodies at the neuromuscular junction. MG is classified by the antigen specificity of these antibodies. Acetylcholine receptor (AChR) antibodies are the most common type (74-88%), followed by anti-muscle specific kinase (MuSK) and other antibodies. While all these antibodies lead to neuromuscular transmission failure, the immuno-pathogenic mechanisms are distinct. Complement activation is a primary driver of AChR antibody-positive MG (AChR+ MG) pathogenesis. This leads to the formation of the membrane attack complex and destruction of AChR receptors and the postsynaptic membrane resulting in impaired neurotransmission and muscle weakness characteristic of MG. Broad-based immune-suppressants like corticosteroids are effective in controlling MG; however, their long-term use can be associated with significant adverse effects. Advances in translational research have led to the development of more directed therapeutic agents that are likely to alter the future of MG treatment. Eculizumab is a humanized monoclonal antibody that inhibits the cleavage of complement protein C5 and is approved for use in generalized MG. In this review, we discuss the pathophysiology of MG; the therapeutic efficacy and tolerability of eculizumab, as well as the practical guidelines for its use in MG; future studies exploring the role of eculizumab in different stages and subtypes of MG subtypes; the optimal duration of therapy and its discontinuation; the characterization of non-responder patients; and the use of biomarkers for monitoring therapy are highlighted. Based on the pathophysiologic mechanisms, emerging therapies and new therapeutic targets are also reviewed.
重症肌无力(MG)是一种罕见的自身免疫性疾病,由神经肌肉接头处的特异性自身抗体引起。MG根据这些抗体的抗原特异性进行分类。乙酰胆碱受体(AChR)抗体是最常见的类型(74 - 88%),其次是抗肌肉特异性激酶(MuSK)抗体和其他抗体。虽然所有这些抗体都会导致神经肌肉传递失败,但免疫致病机制各不相同。补体激活是AChR抗体阳性重症肌无力(AChR + MG)发病机制的主要驱动因素。这会导致膜攻击复合物的形成以及AChR受体和突触后膜的破坏,从而导致神经传递受损和MG特有的肌肉无力。像皮质类固醇这样的广谱免疫抑制剂在控制MG方面有效;然而,长期使用可能会伴有显著的不良反应。转化研究的进展导致了更具针对性的治疗药物的开发,这些药物可能会改变MG治疗的未来。依库珠单抗是一种人源化单克隆抗体,可抑制补体蛋白C5的裂解,已被批准用于全身型MG。在本综述中,我们讨论了MG的病理生理学;依库珠单抗的治疗效果和耐受性,以及其在MG中使用的实用指南;探索依库珠单抗在MG不同阶段和亚型中的作用的未来研究;最佳治疗持续时间及其停药;无反应患者的特征;以及用于监测治疗的生物标志物的使用。基于病理生理机制,还对新兴疗法和新的治疗靶点进行了综述。