Mousa Jehan, Veres Larissa, Mohamed Anab, De Graef Diederik, Morava Eva
Department of Clinical Genomics, Mayo Clinic, Rochester, MN, USA.
Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN, USA.
Mol Genet Metab Rep. 2022 Jul 25;32:100901. doi: 10.1016/j.ymgmr.2022.100901. eCollection 2022 Sep.
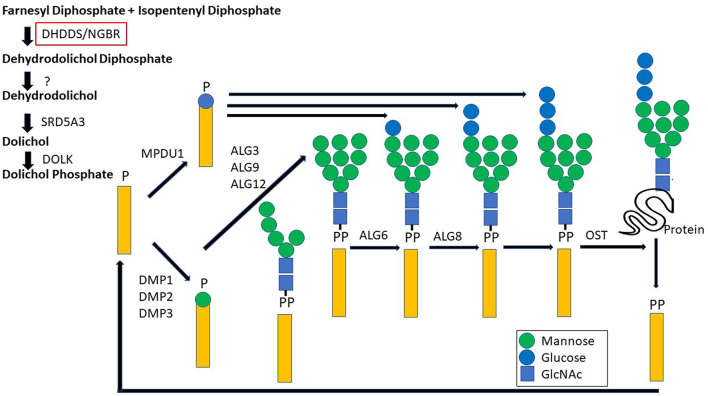
Pathogenic variants in have been associated with either autosomal recessive retinitis pigmentosa or DHDDS-CDG. Heterozygous variants in have been described in patients with a progressive neurodegenerative disease. Here we report on an individual presenting with a multisystem CDG phenotype who was diagnosed with known homozygous pathogenic variants, previously associated with isolated retinitis pigmentosa. An adult Ashkenazi Jewish female developed multiple symptoms of late onset type 1 CDG including seizures, ataxia, protein losing enteropathy, tremor, and titubation in association with elevated mono-oligo/di-oligo transferrin ratio in blood, and classic retinitis pigmentosa. She was diagnosed by whole exome sequencing with the common Ashkenazi Jewish, homozygous p.K42E variants in She was started on Acetazolamide and responded well to the treatment which improved her titubation, tremor, and generalized edema. Reviewing the literature, families with variants and multisystem presentation were different from our patient's presentation in terms of clinical manifestations, severity, genetic defect, and mode of inheritance. In previously reported patients with neurologic symptoms including seizures, movement abnormalities, and global development delay, the phenotype was caused by heterozygous pathogenic variants in . The infant who was reported with a multisystem phenotype and fatal type 1 CDG had compound heterozygosity for a nonsense and a splice site variant in resulting in DHDDS-CDG. The discovery of the novel phenotype associated with the common p.K42E pathogenic variant in expands the spectrum of CDG and further enhances our understanding on the role of in glycosylation beyond the retina.
[基因名称]中的致病变异与常染色体隐性遗传性视网膜色素变性或DHDDS - CDG相关。[基因名称]中的杂合变异已在患有进行性神经退行性疾病的患者中被描述。在此,我们报告一名表现为多系统CDG表型的个体,其被诊断出具有已知的纯合致病[基因名称]变异,该变异先前与孤立性视网膜色素变性相关。一名成年阿什肯纳兹犹太女性出现了迟发型1型CDG的多种症状,包括癫痫发作、共济失调、蛋白丢失性肠病、震颤和躯干共济失调,同时血液中单体寡糖/二聚体寡糖转铁蛋白比率升高,并伴有典型的视网膜色素变性。通过全外显子测序,她被诊断为携带常见的阿什肯纳兹犹太纯合p.K42E变异。她开始使用乙酰唑胺治疗,治疗反应良好,改善了她的躯干共济失调、震颤和全身性水肿。回顾文献,具有[基因名称]变异和多系统表现的家族在临床表现、严重程度、基因缺陷和遗传模式方面与我们患者的表现不同。在先前报道的有癫痫发作、运动异常和全面发育迟缓等神经症状的患者中,表型是由[基因名称]中的杂合致病变异引起的。报道的患有多系统表型和致命性1型CDG的婴儿在[基因名称]中具有一个无义变异和一个剪接位点变异的复合杂合性,导致DHDDS - CDG。与常见的p.K42E致病变异相关的新表型的发现扩展了CDG的谱,并进一步增强了我们对[基因名称]在视网膜以外糖基化作用的理解。