Division of Genetics and Metabolism, Department of Medical Research, MacKay Memorial Hospital, Taipei 10449, Taiwan.
College of Medicine, Fu-Jen Catholic University, New Taipei City 24205, Taiwan.
Int J Mol Sci. 2022 Sep 1;23(17):9979. doi: 10.3390/ijms23179979.
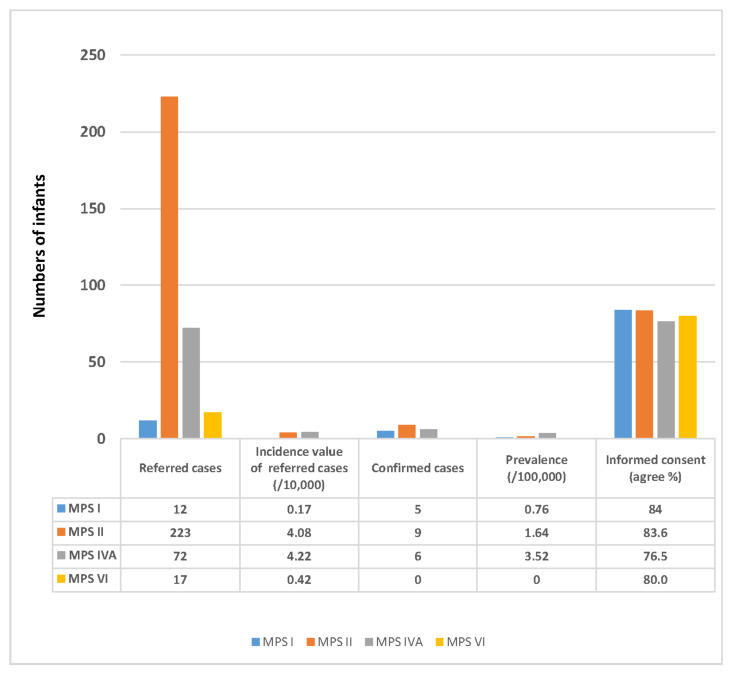
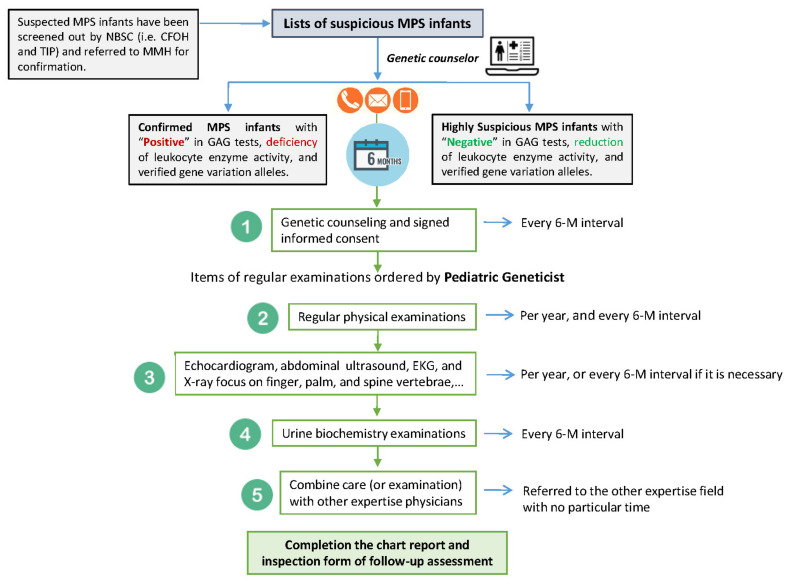
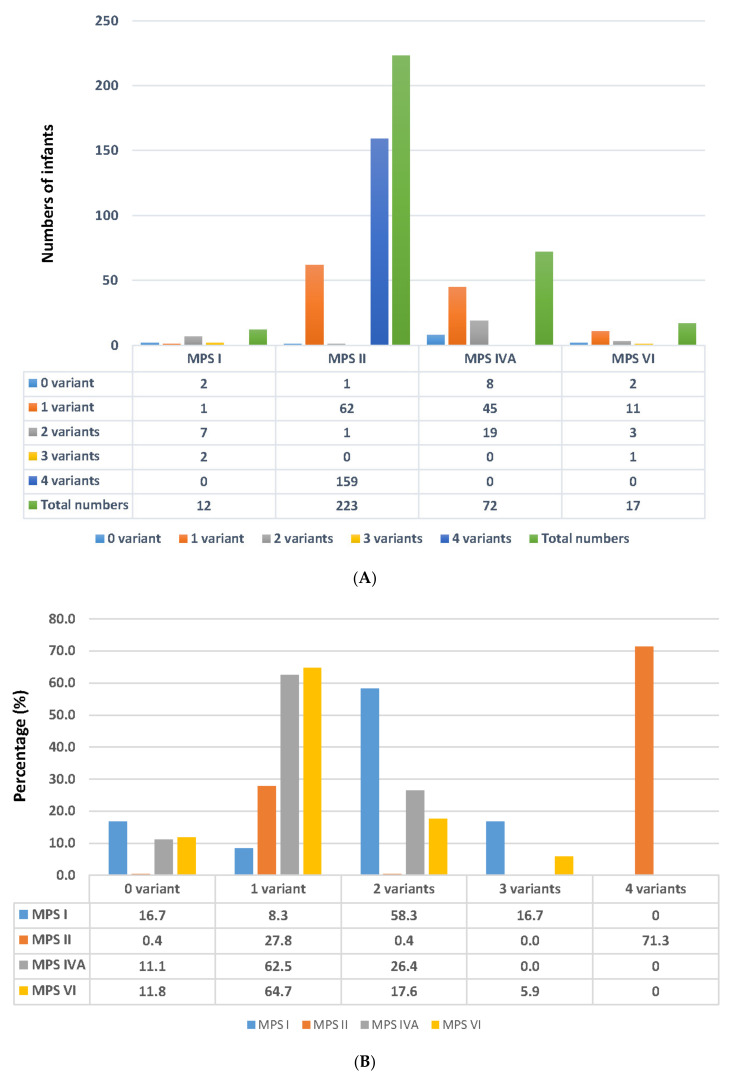
Mucopolysaccharidosis (MPS) is a lysosomal storage disease caused by genetic defects that result in deficiency of one specific enzyme activity, consequently impairing the stepwise degradation of glycosaminoglycans (GAGs). Except for MPS II, the other types of MPS have autosomal recessive inheritance in which two copies of an abnormal allele must be present in order for the disease to develop. In this study, we present the status of variant alleles and biochemistry results found in infants suspected of having MPS I, II, IVA, and VI. A total of 324 suspected infants, including 12 for MPS I, 223 for MPS II, 72 for MPS IVA, and 17 for MPS VI, who were referred for MPS confirmation from newborn screening centers in Taiwan, were enrolled. In all of these infants, one specific enzyme activity in dried blood spot filter paper was lower than the cut-off value in the first blood sample, as well asin a second follow-up sample. The confirmatory methods used in this study included Sanger sequencing, next-generation sequencing, leukocyte enzyme fluorometric assay, and GAG-derived disaccharides in urine using tandem mass spectrometry assays. The results showed that five, nine, and six infants had MPS I, II, and IVA, respectively, and all of them were asymptomatic. Thus, a laboratory diagnosis is extremely important to confirm the diagnosis of MPS. The other infants with identified nucleotide variations and reductions in leukocyte enzyme activities were categorized as being highly suspected cases requiring long-term and intensive follow-up examinations. In summary, the final confirmation of MPS depends on the most powerful biomarkers found in urine, i.e., the quantification of GAG-derived disaccharides including dermatan sulfate, heparan sulfate, and keratan sulfate, and analysis of genetic variants can help predict outcomes and guide treatment.
黏多糖贮积症(MPS)是一种溶酶体贮积病,由遗传缺陷引起,导致一种特定酶活性缺乏,从而使糖胺聚糖(GAG)的逐步降解受损。除 MPS II 外,其他类型的 MPS 为常染色体隐性遗传,即必须存在两个异常等位基因的拷贝,疾病才会发展。在这项研究中,我们介绍了疑似患有 MPS I、II、IVA 和 VI 的婴儿的变异等位基因和生化结果的状况。共有 324 名疑似婴儿,包括 12 名 MPS I、223 名 MPS II、72 名 MPS IVA 和 17 名 MPS VI,他们均因在台湾新生儿筛查中心进行的 MPS 确认而被转诊。在所有这些婴儿中,干血斑滤纸中的一种特定酶活性均低于第一份血样和第二份随访血样的截止值。本研究中使用的确认方法包括 Sanger 测序、下一代测序、白细胞酶荧光测定法以及串联质谱法测定尿中 GAG 衍生的二糖。结果表明,有五名、九名和六名婴儿分别患有 MPS I、II 和 IVA,且均无症状。因此,实验室诊断对于确认 MPS 诊断极为重要。其他具有核苷酸变异和白细胞酶活性降低的婴儿被归类为高度疑似病例,需要长期和强化的随访检查。总之,MPS 的最终确认取决于尿液中发现的最有力的生物标志物,即定量分析包括硫酸皮肤素、硫酸乙酰肝素和硫酸角质素在内的 GAG 衍生二糖,以及遗传变异分析有助于预测结果并指导治疗。