Division of Pulmonary, Critical Care and Sleep Medicine, University of Cincinnati, Cincinnati, OH 45267-0564, USA.
Int J Mol Sci. 2023 Feb 2;24(3):2850. doi: 10.3390/ijms24032850.
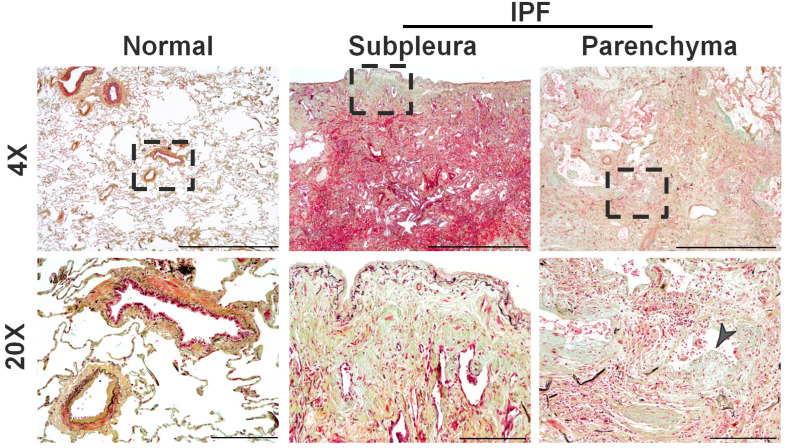
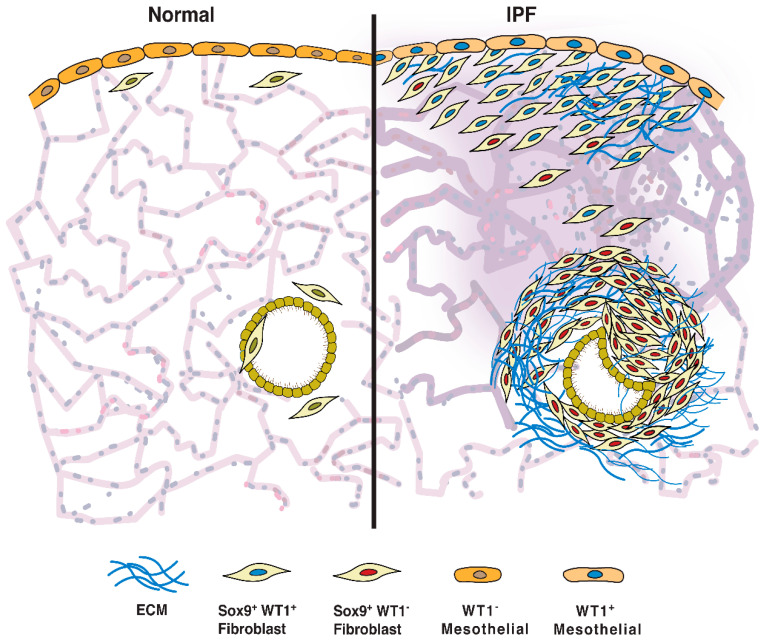
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic lung disease that is often fatal due to the formation of irreversible scar tissue in the distal areas of the lung. Although the pathological and radiological features of IPF lungs are well defined, the lack of insight into the fibrogenic role of fibroblasts that accumulate in distinct anatomical regions of the lungs is a critical knowledge gap. Fibrotic lesions have been shown to originate in the subpleural areas and extend into the lung parenchyma through processes of dysregulated fibroproliferation, migration, fibroblast-to-myofibroblast transformation, and extracellular matrix production. Identifying the molecular targets underlying subpleural thickening at the early and late stages of fibrosis could facilitate the development of new therapies to attenuate fibroblast activation and improve the survival of patients with IPF. Here, we discuss the key cellular and molecular events that contribute to (myo)fibroblast activation and subpleural thickening in IPF. In particular, we highlight the transcriptional programs involved in mesothelial to mesenchymal transformation and fibroblast dysfunction that can be targeted to alter the course of the progressive expansion of fibrotic lesions in the distal areas of IPF lungs.
特发性肺纤维化(IPF)是一种进行性肺纤维化疾病,由于远端肺组织中不可逆的瘢痕组织形成,常导致死亡。尽管 IPF 肺部的病理和放射学特征已经明确,但对于积聚在肺部不同解剖区域的成纤维细胞的纤维化作用缺乏深入了解,这是一个关键的知识空白。纤维化病变已被证明起源于胸膜下区域,并通过失调的纤维增生、迁移、成纤维细胞向肌成纤维细胞转化和细胞外基质产生等过程扩展到肺实质。确定纤维化早期和晚期胸膜下增厚的分子靶点,可以促进开发新的治疗方法来减弱成纤维细胞的激活,并提高 IPF 患者的生存率。在这里,我们讨论了导致(肌)成纤维细胞激活和 IPF 胸膜下增厚的关键细胞和分子事件。特别是,我们强调了参与间皮到间充质转化和成纤维细胞功能障碍的转录程序,这些程序可以作为靶点,改变 IPF 肺部远端纤维化病变进行性扩展的过程。