Boncompagni Alessandra, Lucas-Herald Angela K, Beattie Paula, McDevitt Helen, Iughetti Lorenzo, Constantinou Panayiotis, Kinning Esther, Ahmed S Faisal, Mason Avril
Developmental Endocrinology Research Group, Royal Hospital for Children, University of Glasgow, Glasgow, United Kingdom.
Postgraduate School of Paediatrics, Department of Medical and Surgical Sciences of Mothers, Children and Adults, University of Modena & Reggio Emilia, Paediatric Unit, Modena, Italy.
Bone Rep. 2023 Feb 23;18:101665. doi: 10.1016/j.bonr.2023.101665. eCollection 2023 Jun.
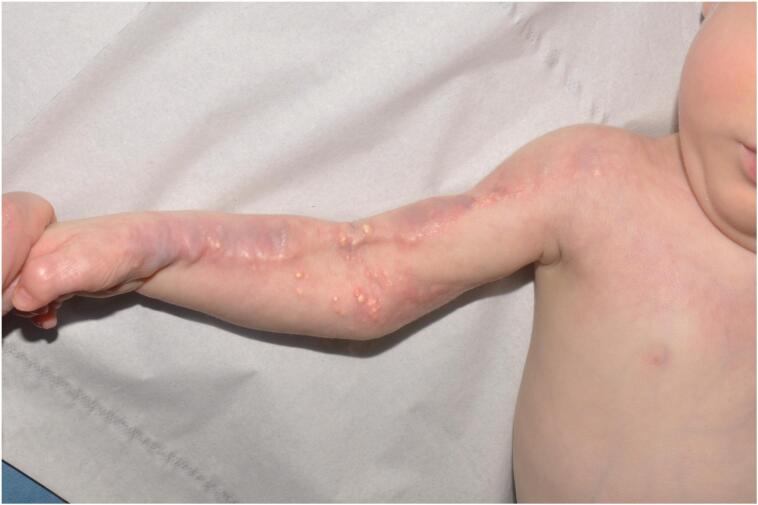
Progressive osseous heteroplasia (POH) is a rare genetic disorder characterised by progressive heterotopic ossification (HO) within the skin and subcutaneous tissues. The condition is caused by heterozygous inactivating mutations of the gene and usually presents in infancy. We describe the case of a white male ex-preterm who was first referred because of subcutaneous calcium deposits along the right arm after extravasation of parenteral nutrition. As these lesions progressed, a skin biopsy was undertaken which revealed intramembranous ossification. Genetic testing revealed a constitutional, , heterozygous, nonsense variant in the gene that has not previously been described, but which is consistent with patient's clinical diagnosis of POH. No endocrine abnormalities or other signs congruent with overlapping conditions were detected. To the best of our knowledge, this is the first case describing an inflammatory trigger in POH. Trials with intravenous bisphosphonate and glucocorticoid as well as with topical sodium thiosulphate were attempted without clinical improvement. Excision of the calcifications and physiotherapy seem to have provided a partial improvement on mobility of the elbow. This case widens the spectrum of phenotypes seen in mutation disorders and suggests that alternative anti-inflammatory treatments may be effective. Mutations in should be considered in cases of significant progressive calcium deposition after extravasation injury.
进行性骨化性纤维发育不良(POH)是一种罕见的遗传性疾病,其特征为皮肤和皮下组织内出现进行性异位骨化(HO)。该病症由该基因的杂合失活突变引起,通常在婴儿期出现。我们描述了一名白人男性前早产儿的病例,他最初因肠外营养渗漏后右臂出现皮下钙沉积而被转诊。随着这些病变的进展,进行了皮肤活检,结果显示为膜内骨化。基因检测发现该基因存在一种此前未被描述的、符合患者POH临床诊断的、组成性的、杂合的无义变异。未检测到内分泌异常或与重叠病症相符的其他体征。据我们所知,这是首例描述POH炎症触发因素的病例。尝试了静脉注射双膦酸盐和糖皮质激素以及局部使用硫代硫酸钠的试验,但均未取得临床改善。钙化切除术和物理治疗似乎对肘部活动度有部分改善。该病例拓宽了在该基因突变疾病中所见的表型谱,并表明替代抗炎治疗可能有效。在发生渗漏损伤后出现明显进行性钙沉积的病例中,应考虑该基因的突变。