Horae Gene Therapy Center.
Department of Neurology.
JCI Insight. 2023 May 8;8(9):e168688. doi: 10.1172/jci.insight.168688.
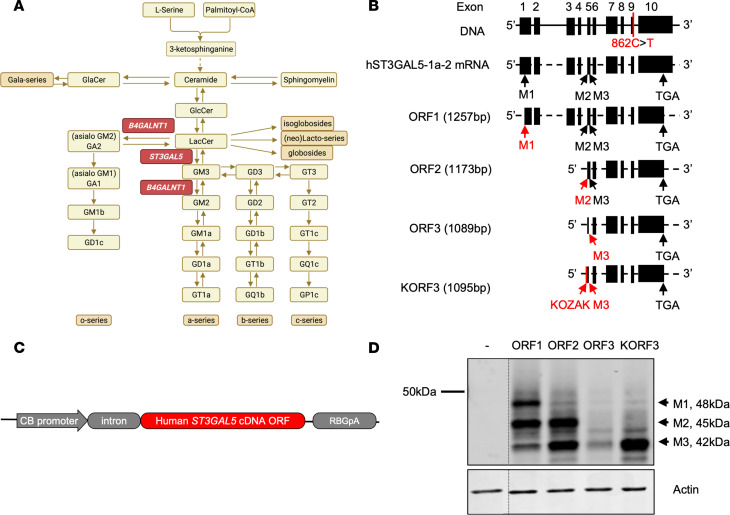
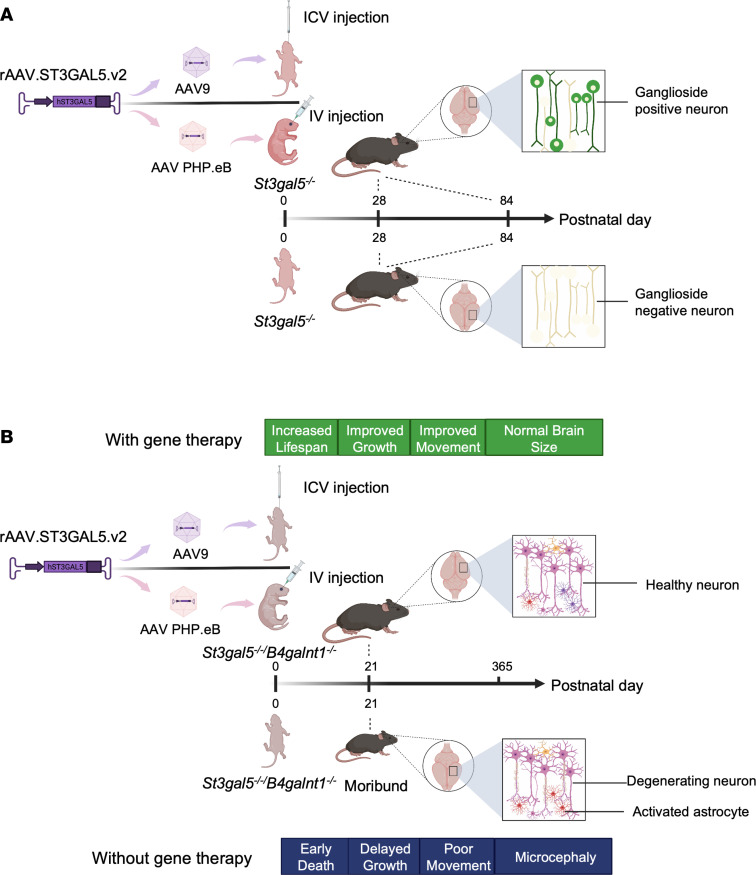
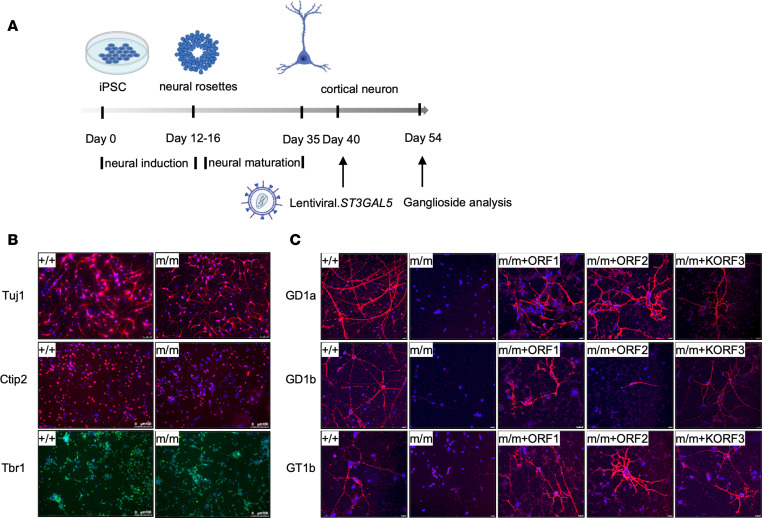
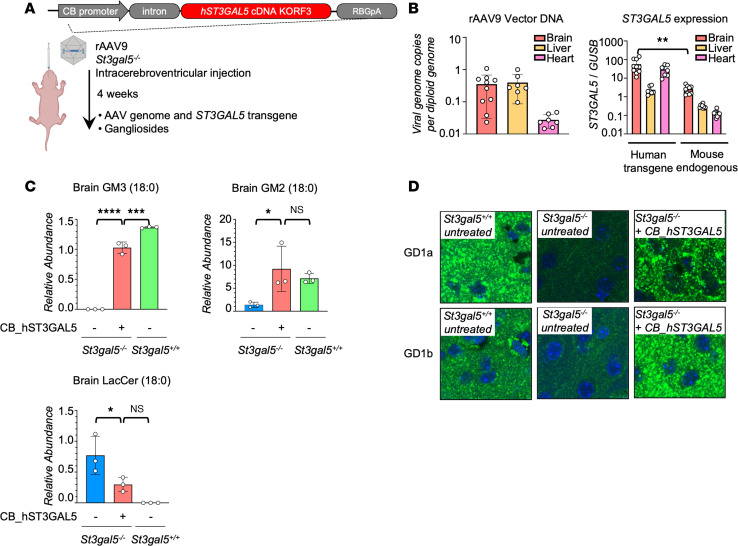
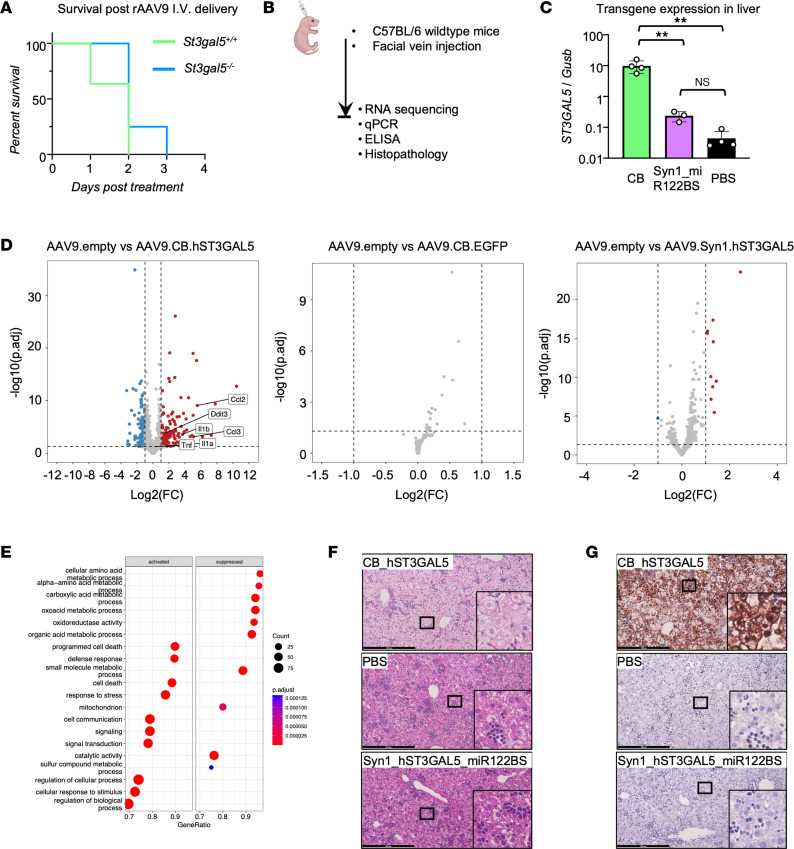
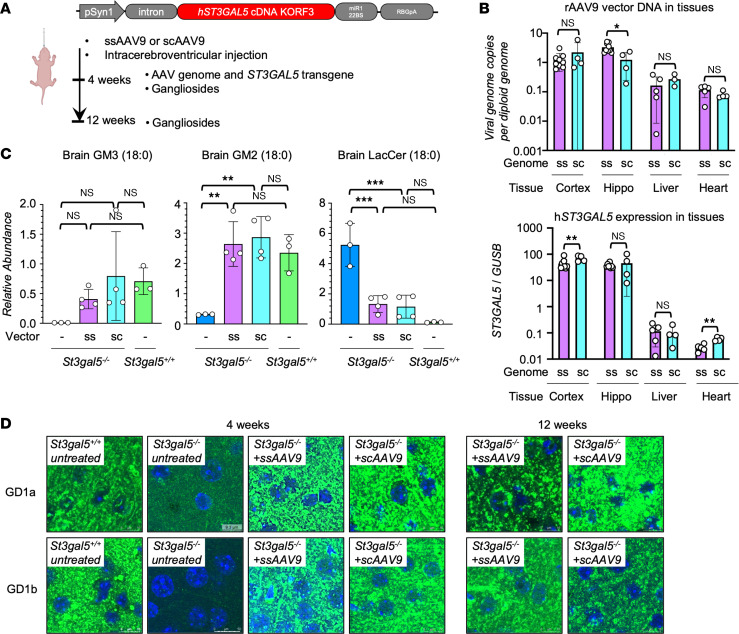
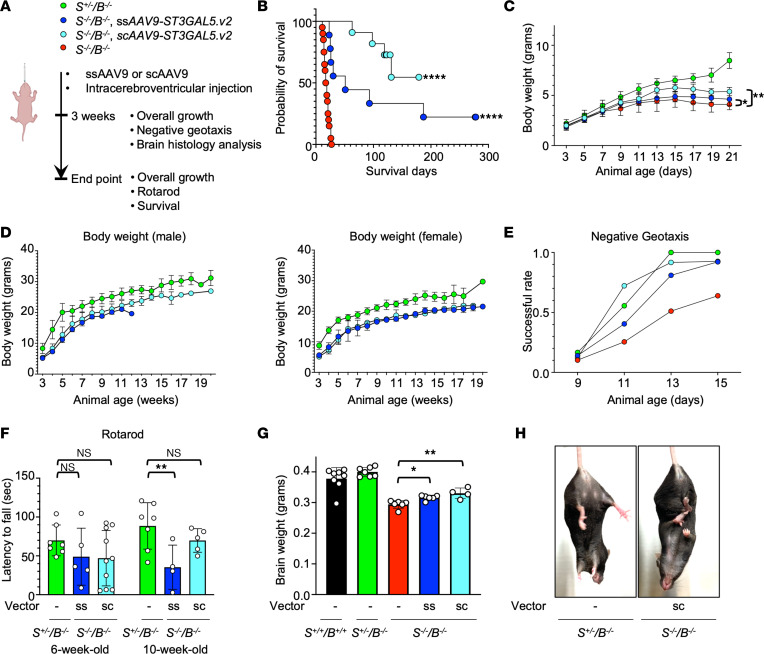
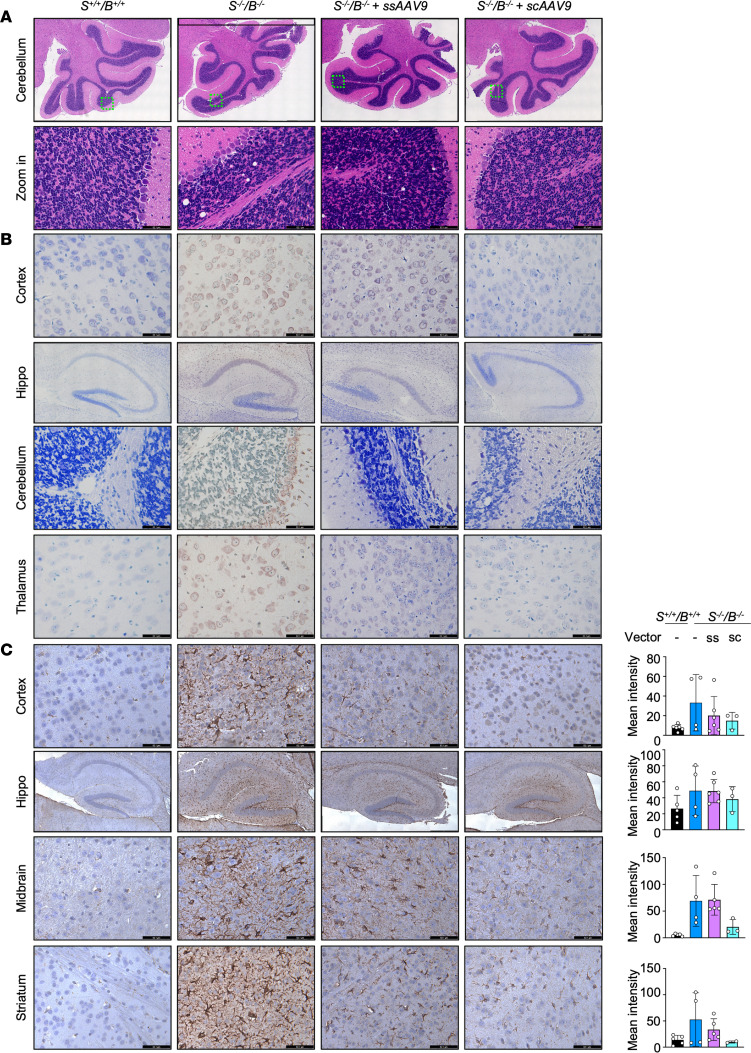
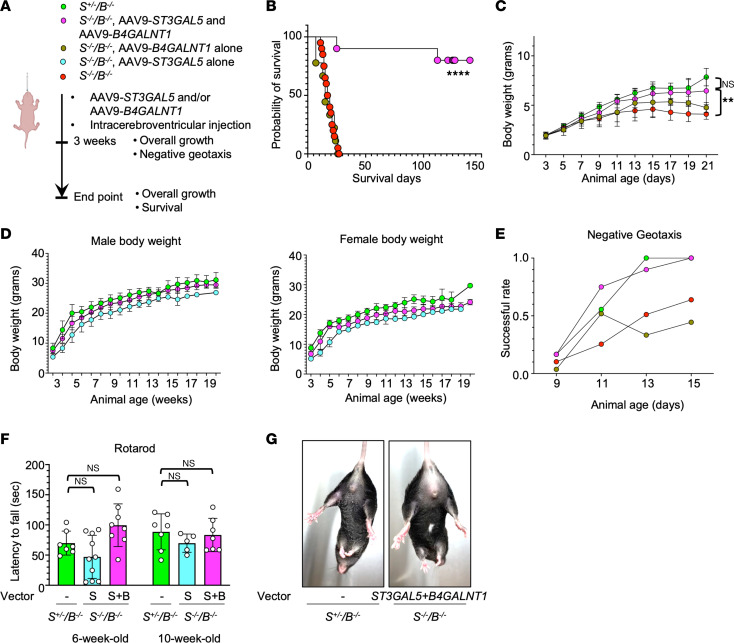
GM3 synthase deficiency (GM3SD) is an infantile-onset epileptic encephalopathy syndrome caused by biallelic loss-of-function mutations in ST3GAL5. Loss of ST3GAL5 activity in humans results in systemic ganglioside deficiency and severe neurological impairment. No disease-modifying treatment is currently available. Certain recombinant adeno-associated viruses (rAAVs) can cross the blood-brain barrier to induce widespread, long-term gene expression in the CNS and represent a promising therapeutic strategy. Here, we show that a first-generation rAAV-ST3GAL5 replacement vector using a ubiquitous promoter restored tissue ST3GAL5 expression and normalized cerebral gangliosides in patient-derived induced pluripotent stem cell neurons and brain tissue from St3gal5-KO mice but caused fatal hepatotoxicity when administered systemically. In contrast, a second-generation vector optimized for CNS-restricted ST3GAL5 expression, administered by either the intracerebroventricular or i.v. route at P1, allowed for safe and effective rescue of lethality and behavior impairment in symptomatic GM3SD mice up to a year. These results support further clinical development of ST3GAL5 gene therapy.
GM3 合酶缺乏症(GM3SD)是一种婴儿期发病的癫痫性脑病综合征,由 ST3GAL5 的双等位基因功能丧失突变引起。人类 ST3GAL5 活性丧失导致全身神经节苷脂缺乏和严重的神经功能障碍。目前尚无疾病修饰治疗方法。某些重组腺相关病毒(rAAV)可以穿透血脑屏障,在中枢神经系统中诱导广泛的长期基因表达,这代表了一种有前途的治疗策略。在这里,我们表明,使用通用启动子的第一代 rAAV-ST3GAL5 替代载体恢复了组织 ST3GAL5 表达,并使患者来源的诱导多能干细胞神经元和 St3gal5-KO 小鼠脑组织中的脑苷脂正常化,但全身性给药时会导致致命的肝毒性。相比之下,在 P1 时通过侧脑室或静脉内途径给药的优化用于中枢神经系统限制的 ST3GAL5 表达的第二代载体允许安全有效地挽救有症状的 GM3SD 小鼠的致死性和行为障碍,最长可达一年。这些结果支持 ST3GAL5 基因治疗的进一步临床开发。