Institute for Neurosciences of Montpellier (INM), University of Montpellier, INSERM, Montpellier, France.
National Reference Centre for Inherited Sensory Diseases, University of Montpellier, CHU, Montpellier, France.
HGG Adv. 2023 Aug 7;4(4):100229. doi: 10.1016/j.xhgg.2023.100229. eCollection 2023 Oct 12.
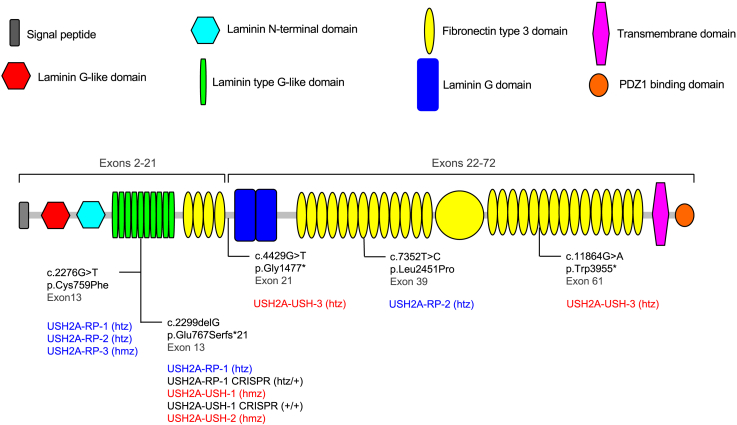
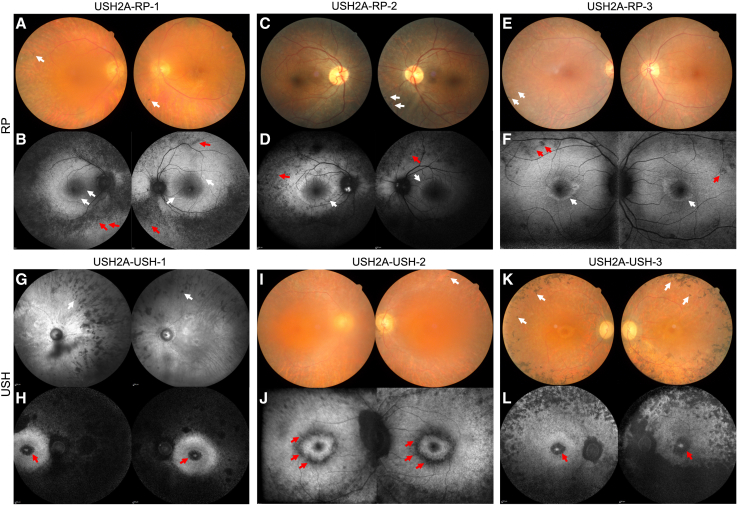
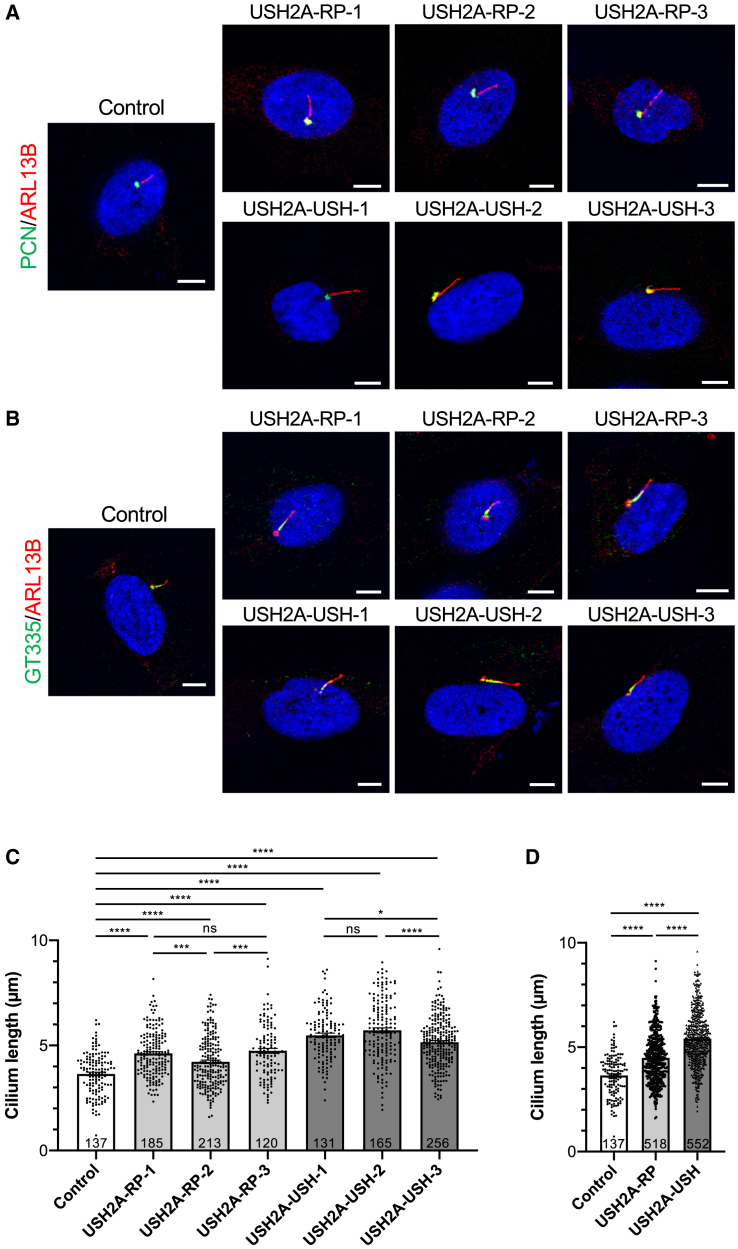
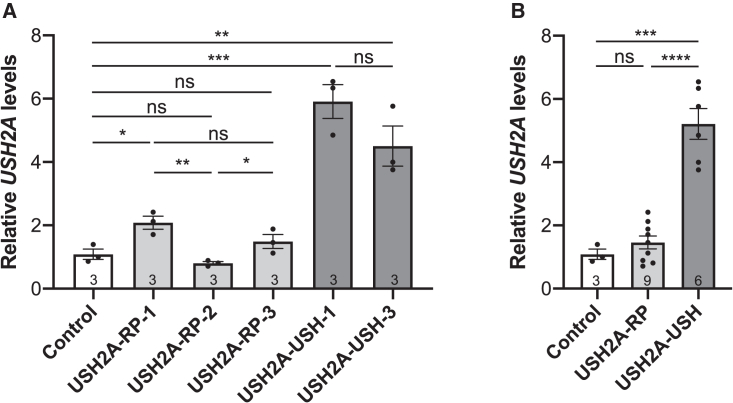
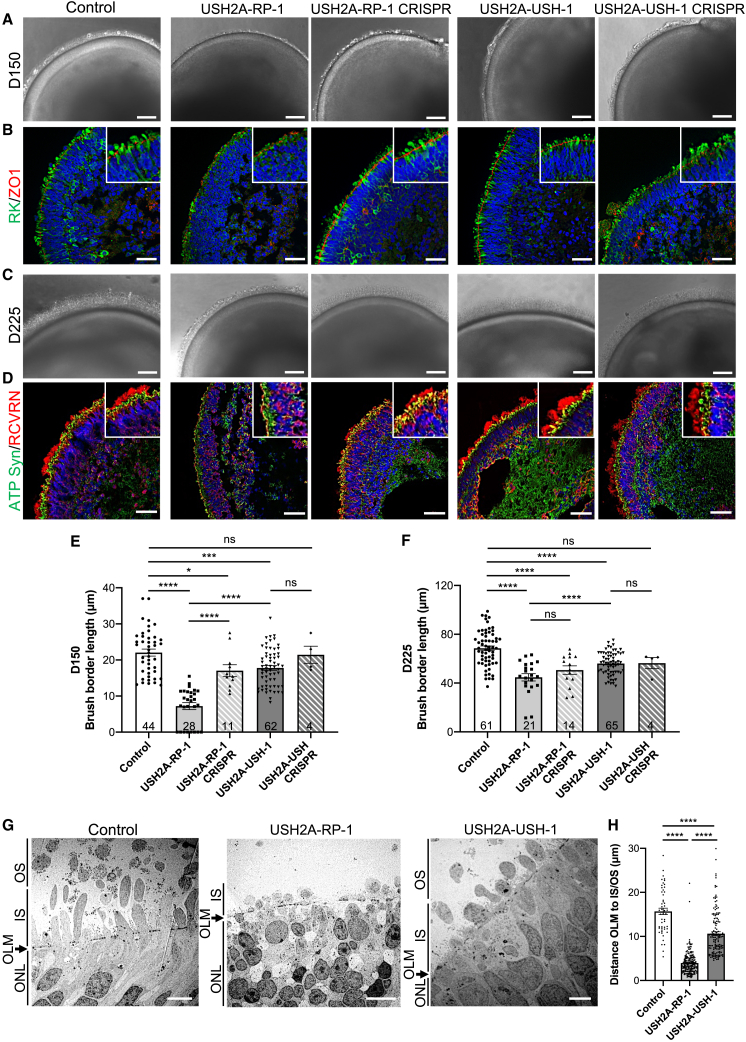
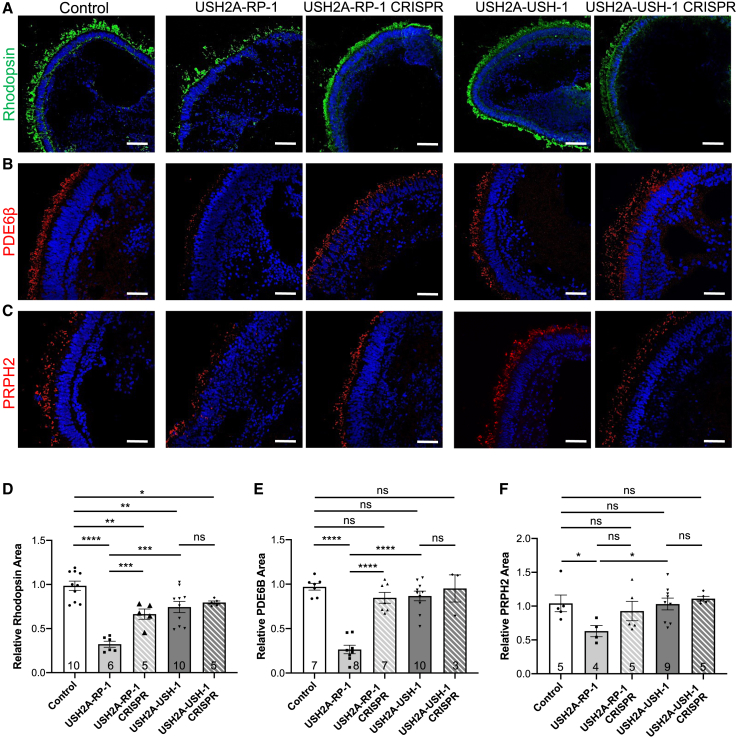
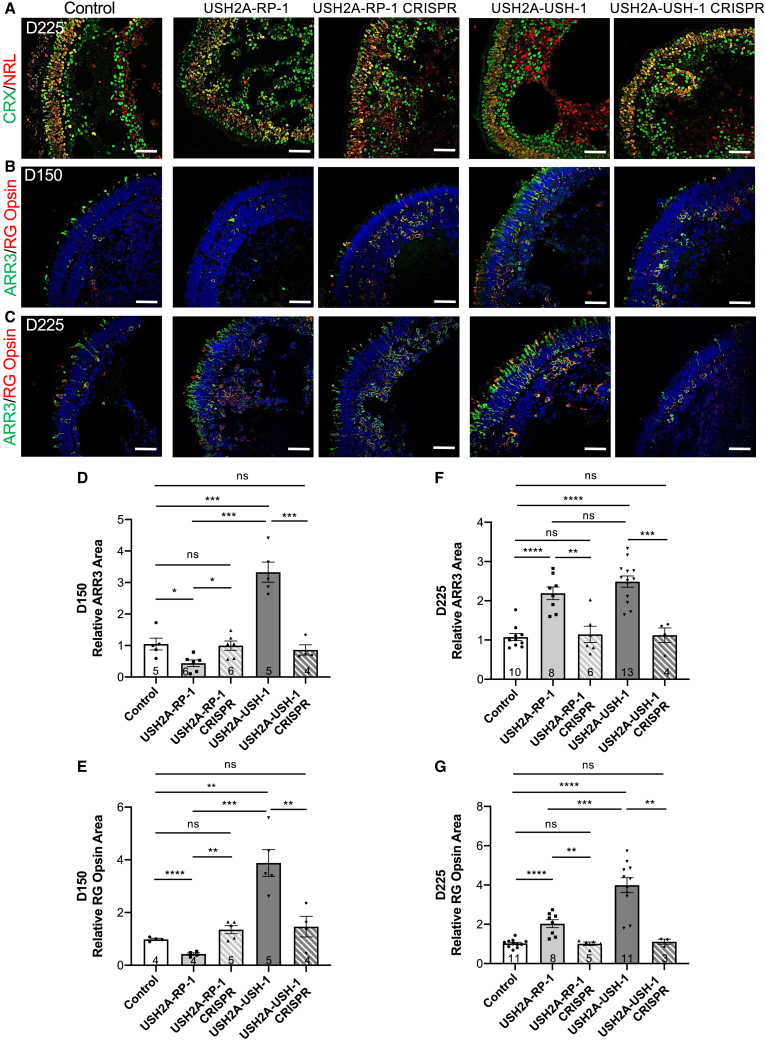
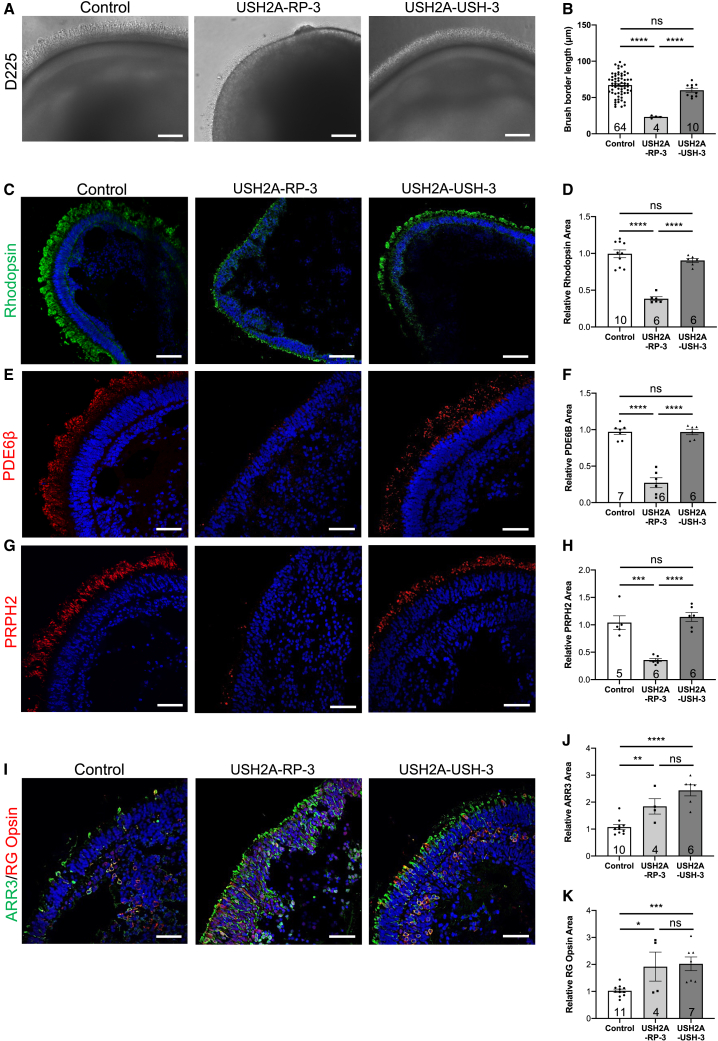
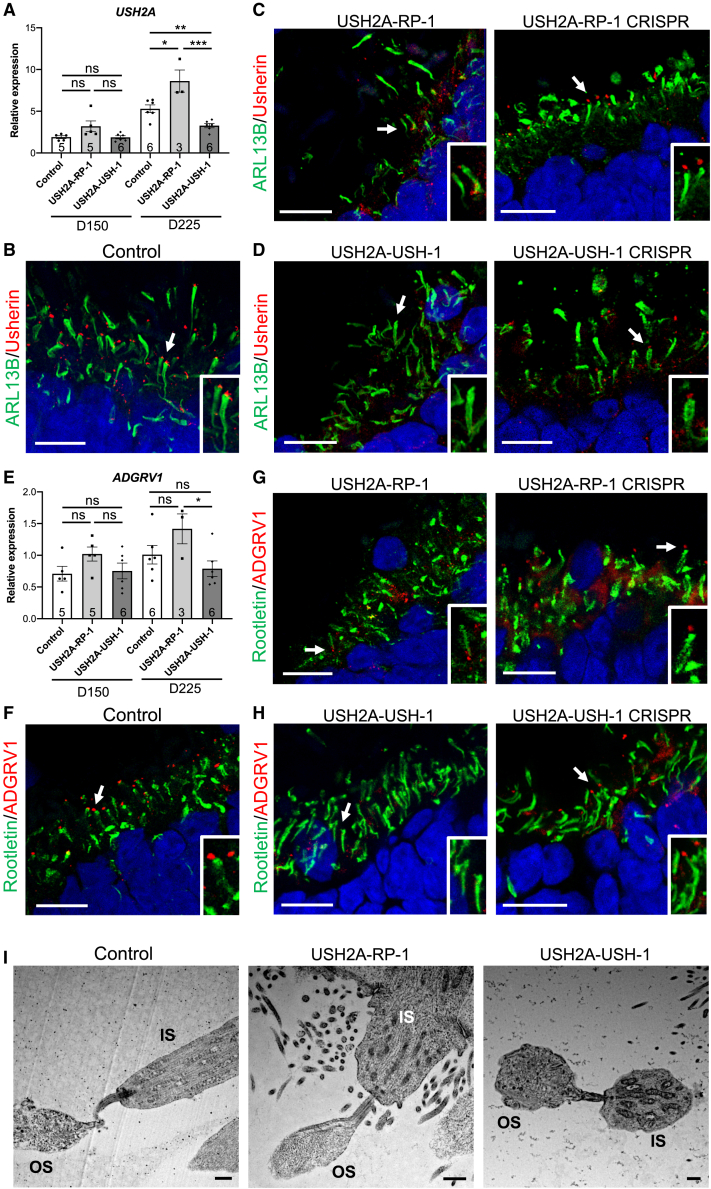
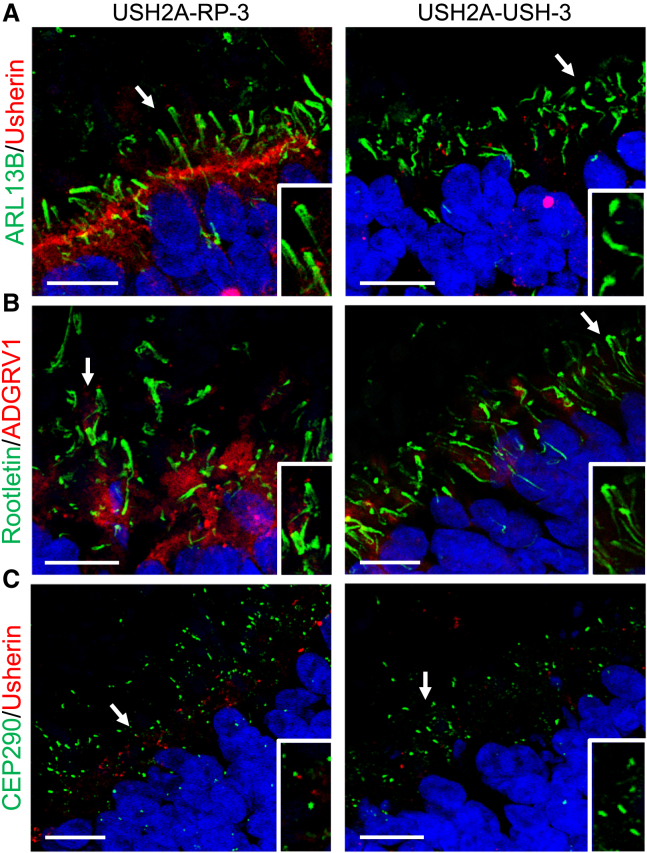
There is an emblematic clinical and genetic heterogeneity associated with inherited retinal diseases (IRDs). The most common form is retinitis pigmentosa (RP), a rod-cone dystrophy caused by pathogenic variants in over 80 different genes. Further complexifying diagnosis, different variants in individual RP genes can also alter the clinical phenotype. is the most prevalent gene for autosomal-recessive RP and one of the most challenging because of its large size and, hence, large number of variants. Moreover, variants give rise to non-syndromic and syndromic RP, known as Usher syndrome (USH) type 2, which is associated with vision and hearing loss. The lack of a clear genotype-phenotype correlation or prognostic models renders diagnosis highly challenging. We report here a long-awaited differential non-syndromic RP and USH phenotype in three human disease-specific models: fibroblasts, induced pluripotent stem cells (iPSCs), and mature iPSC-derived retinal organoids. Moreover, we identified distinct retinal phenotypes in organoids from multiple RP and USH individuals, which were validated by isogenic-corrected controls. Non-syndromic RP organoids showed compromised photoreceptor differentiation, whereas USH organoids showed a striking and unexpected cone phenotype. Furthermore, complementary clinical investigations identified macular atrophy in a high proportion of USH compared with RP individuals, further validating our observations that variants differentially affect cones. Overall, identification of distinct non-syndromic RP and USH phenotypes in multiple models provides valuable and robust readouts for testing the pathogenicity of variants as well as the efficacy of therapeutic approaches in complementary cell types.
遗传性视网膜疾病(IRDs)具有明显的临床和遗传异质性。最常见的形式是色素性视网膜炎(RP),这是一种由 80 多种不同基因的致病变体引起的视杆-视锥营养不良。进一步使诊断复杂化的是,个体 RP 基因中的不同变体也可以改变临床表型。 是常染色体隐性 RP 最常见的基因,也是最具挑战性的基因之一,因为其体积大,因此变体数量也多。此外, 变体导致非综合征和综合征 RP,称为 Usher 综合征(USH)2 型,其与视力和听力损失有关。由于缺乏明确的基因型-表型相关性或预测模型,因此诊断极具挑战性。我们在此报告了在三种人类疾病特异性模型(成纤维细胞、诱导多能干细胞(iPSC)和成熟的 iPSC 衍生的视网膜类器官)中期待已久的非综合征性 RP 和 USH 表型差异。此外,我们在多个 RP 和 USH 个体的类器官中鉴定出了不同的视网膜表型,这些表型通过同源校正对照得到了验证。非综合征性 RP 类器官显示感光细胞分化受损,而 USH 类器官显示出惊人且出乎意料的锥细胞表型。此外,补充临床研究发现,与 RP 个体相比,USH 个体中黄斑萎缩的比例更高,进一步验证了我们的观察结果,即 变体差异地影响锥细胞。总体而言,在多种模型中鉴定出不同的非综合征性 RP 和 USH 表型,为测试 变体的致病性以及互补细胞类型中治疗方法的疗效提供了有价值且稳健的检测结果。