Solanki Kaushal V, Hu Yirui, Moore Bryn S, Abedi Vida, Avula Venkatesh, Mirshahi Tooraj, Strande Natasha T, Bucaloiu Ion D, Chang Alexander R
Center for Kidney Health Research, Geisinger, Danville, Pennsylvania, USA.
Department of Population Health Sciences, Geisinger, Danville, Pennsylvania, USA.
Kidney Int Rep. 2023 Jul 25;8(10):2088-2099. doi: 10.1016/j.ekir.2023.07.010. eCollection 2023 Oct.
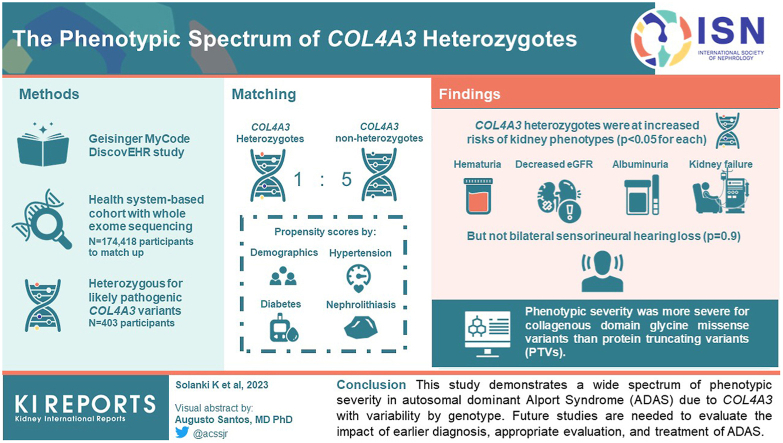
The penetrance and phenotypic spectrum of autosomal dominant Alport Syndrome (ADAS), affecting 1 in 106, remains understudied.
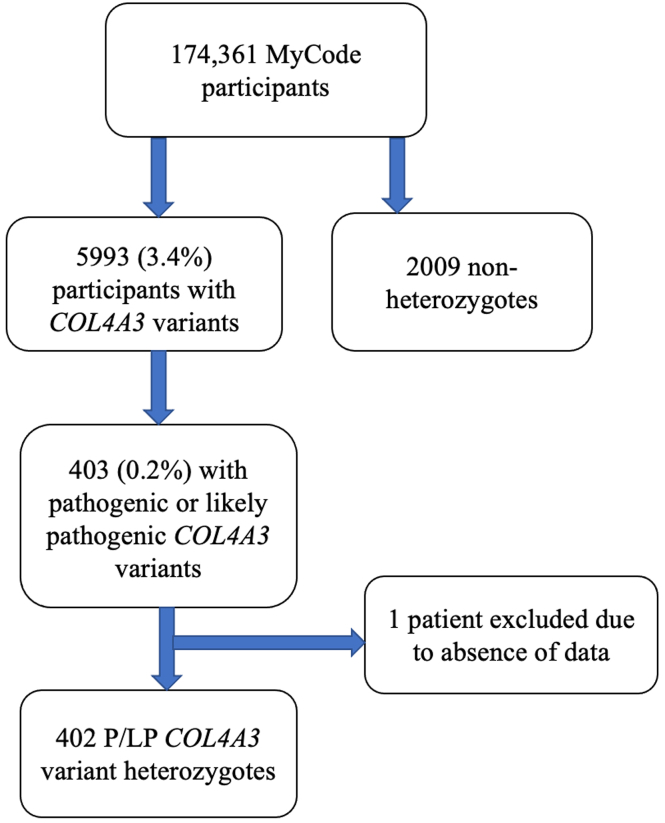
Using data from 174,418 participants in the Geisinger MyCode/DiscovEHR study, an unselected health system-based cohort with whole exome sequencing, we identified 403 participants who were heterozygous for likely pathogenic variants. Phenotypic data was evaluated using International Classification of Diseases (ICD) codes, laboratory data, and chart review. To evaluate the phenotypic spectrum of genetically-determined ADAS, we matched heterozygotes 1:5 to nonheterozygotes using propensity scores by demographics, hypertension, diabetes, and nephrolithiasis.
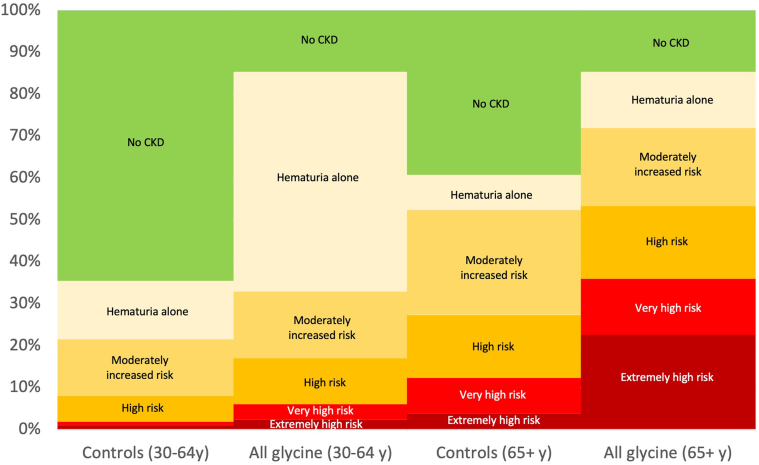
heterozygotes were at significantly increased risks of hematuria, decreased estimated glomerular filtration rate (eGFR), albuminuria, and kidney failure ( < 0.05 for all comparisons) but not bilateral sensorineural hearing loss ( = 0.9). Phenotypic severity was more severe for collagenous domain glycine missense variants than protein truncating variants (PTVs). For example, patients with Gly695Arg ( = 161) had markedly increased risk of dipstick hematuria (odds ratio [OR] 9.50; 95% confidence interval [CI]: 6.32, 14.28) and kidney failure (OR 7.02; 95% CI: 3.48, 14.16) whereas those with PTVs ( = 119) had moderately increased risks of dipstick hematuria (OR 1.64; 95% CI: 1.03, 2.59) and kidney failure (OR 3.44; 95% CI: 1.28, 9.22). Less than a third of patients had albuminuria screening completed, and fewer than 1 of 3 were taking inhibitors of the renin-angiotensin-aldosterone system.
This study demonstrates a wide spectrum of phenotypic severity in ADAS due to with phenotypic variability by genotype. Future studies are needed to evaluate the impact of earlier diagnosis, appropriate evaluation, and treatment of ADAS.
常染色体显性遗传性阿尔波特综合征(ADAS)的外显率和表型谱影响着1/106的人群,目前仍研究不足。
利用盖辛格MyCode/DiscovEHR研究中174418名参与者的数据,这是一个基于健康系统的未选择队列,进行了全外显子测序,我们确定了403名携带可能致病变异的杂合子参与者。使用国际疾病分类(ICD)编码、实验室数据和病历审查来评估表型数据。为了评估基因决定的ADAS的表型谱,我们通过人口统计学、高血压、糖尿病和肾结石的倾向评分,将杂合子与非杂合子按1:5进行匹配。
杂合子出现血尿、估计肾小球滤过率(eGFR)降低、蛋白尿和肾衰竭的风险显著增加(所有比较P<0.05),但双侧感音神经性听力损失风险未增加(P = 0.9)。胶原结构域甘氨酸错义变异的表型严重程度比蛋白质截短变异(PTV)更严重。例如,携带Gly695Arg变异(n = 161)的患者出现试纸法血尿(优势比[OR]9.50;95%置信区间[CI]:6.32,14.28)和肾衰竭(OR 7.02;95%CI:3.48,14.16)的风险显著增加,而携带PTV变异(n = 119)的患者出现试纸法血尿(OR 1.64;95%CI:1.03,2.59)和肾衰竭(OR 3.44;95%CI:1.28,9.22)的风险适度增加。不到三分之一的患者完成了蛋白尿筛查,三分之一的患者中服用肾素-血管紧张素-醛固酮系统抑制剂的不到一人。
本研究表明,ADAS存在广泛的表型严重程度,且因基因型而异。未来需要开展研究,以评估ADAS早期诊断、适当评估和治疗的影响。