Sidpra Jai, Sudhakar Sniya, Biswas Asthik, Massey Flavia, Turchetti Valentina, Lau Tracy, Cook Edward, Alvi Javeria Raza, Elbendary Hasnaa M, Jewell Jerry L, Riva Antonella, Orsini Alessandro, Vignoli Aglaia, Federico Zara, Rosenblum Jessica, Schoonjans An-Sofie, de Wachter Matthias, Delgado Alvarez Ignacio, Felipe-Rucián Ana, Haridy Nourelhoda A, Haider Shahzad, Zaman Mashaya, Banu Selina, Anwaar Najwa, Rahman Fatima, Maqbool Shazia, Yadav Rashmi, Salpietro Vincenzo, Maroofian Reza, Patel Rajan, Radhakrishnan Rupa, Prabhu Sanjay P, Lichtenbelt Klaske, Stewart Helen, Murakami Yoshiko, Löbel Ulrike, D'Arco Felice, Wakeling Emma, Jones Wendy, Hay Eleanor, Bhate Sanjay, Jacques Thomas S, Mirsky David M, Whitehead Matthew T, Zaki Maha S, Sultan Tipu, Striano Pasquale, Jansen Anna C, Lequin Maarten, de Vries Linda S, Severino Mariasavina, Edmondson Andrew C, Menzies Lara, Campeau Philippe M, Houlden Henry, McTague Amy, Efthymiou Stephanie, Mankad Kshitij
Developmental Biology and Cancer Section, University College London Great Ormond Street Institute of Child Health, London WC1N 1EH, UK.
Department of Neuroradiology, Great Ormond Street Hospital for Children NHS Foundation Trust, London WC1N 3JH, UK.
Brain. 2024 Aug 1;147(8):2775-2790. doi: 10.1093/brain/awae056.
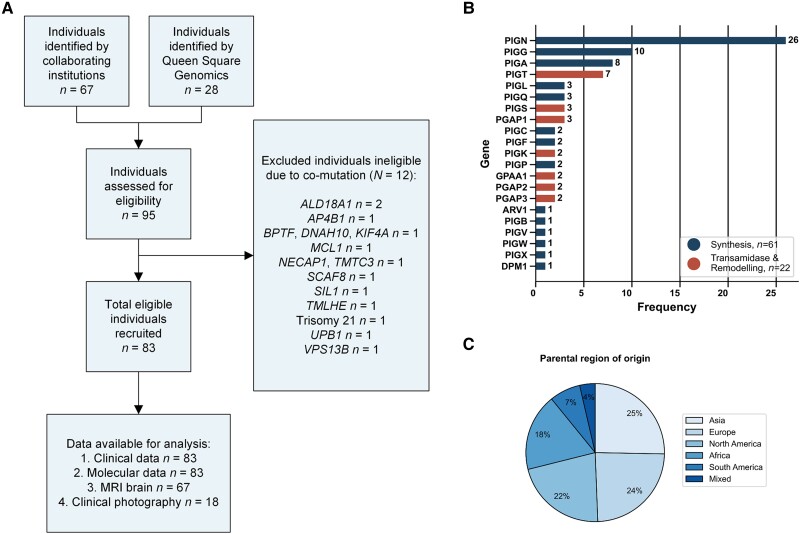
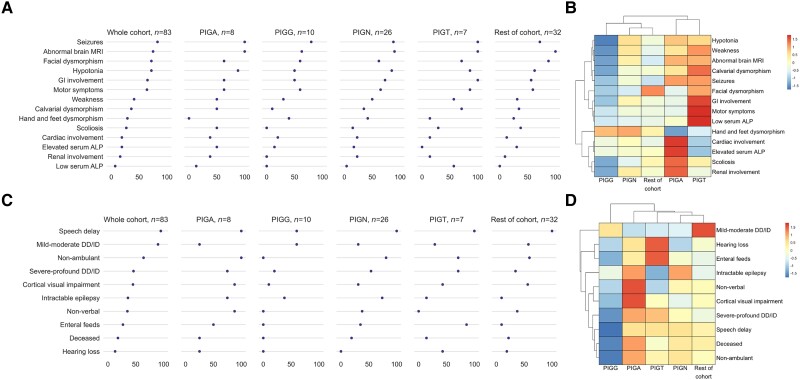
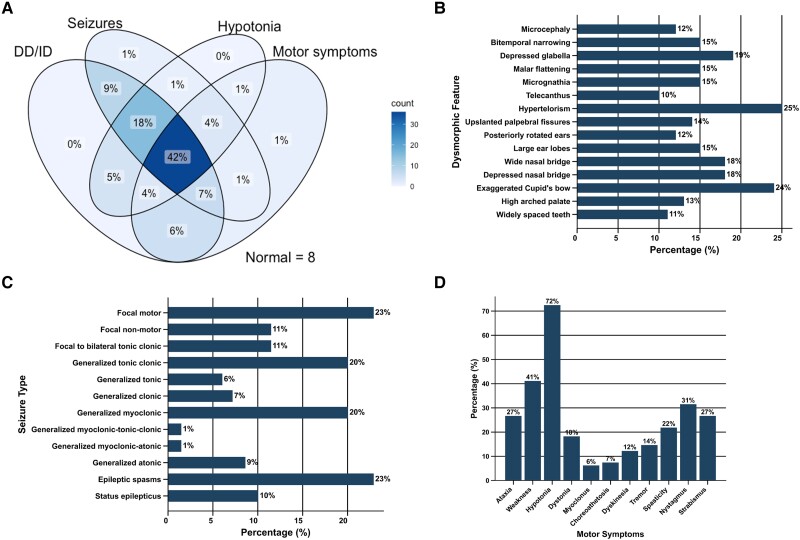
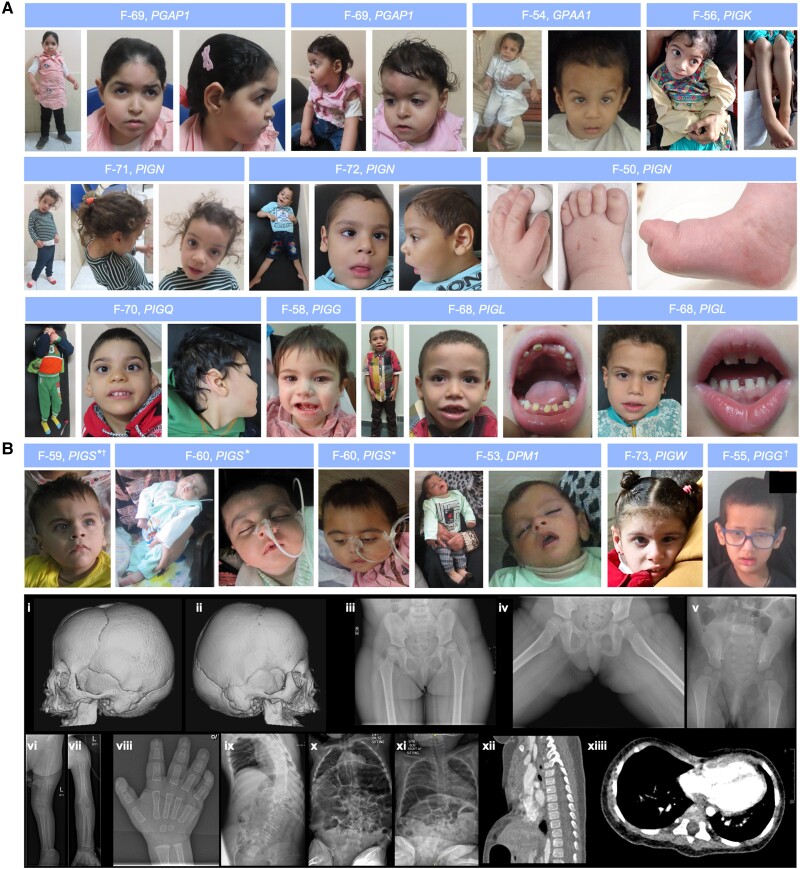
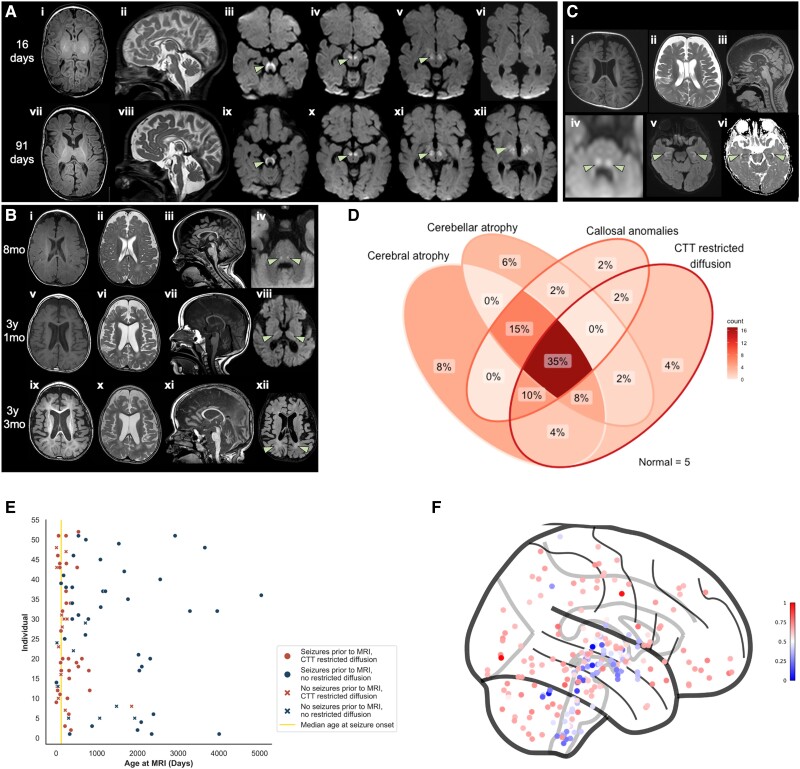
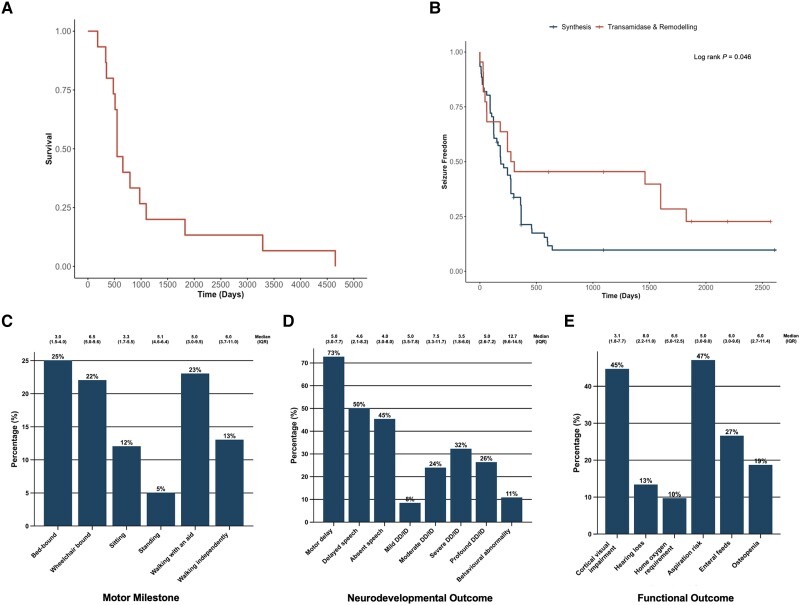
Inherited glycosylphosphatidylinositol deficiency disorders (IGDs) are a group of rare multisystem disorders arising from pathogenic variants in glycosylphosphatidylinositol anchor pathway (GPI-AP) genes. Despite associating 24 of at least 31 GPI-AP genes with human neurogenetic disease, prior reports are limited to single genes without consideration of the GPI-AP as a whole and with limited natural history data. In this multinational retrospective observational study, we systematically analyse the molecular spectrum, phenotypic characteristics and natural history of 83 individuals from 75 unique families with IGDs, including 70 newly reported individuals; the largest single cohort to date. Core clinical features were developmental delay or intellectual disability (DD/ID, 90%), seizures (83%), hypotonia (72%) and motor symptoms (64%). Prognostic and biologically significant neuroimaging features included cerebral atrophy (75%), cerebellar atrophy (60%), callosal anomalies (57%) and symmetric restricted diffusion of the central tegmental tracts (60%). Sixty-one individuals had multisystem involvement including gastrointestinal (66%), cardiac (19%) and renal (14%) anomalies. Though dysmorphic features were appreciated in 82%, no single dysmorphic feature had a prevalence >30%, indicating substantial phenotypic heterogeneity. Follow-up data were available for all individuals, 15 of whom were deceased at the time of writing. Median age at seizure onset was 6 months. Individuals with variants in synthesis stage genes of the GPI-AP exhibited a significantly shorter time to seizure onset than individuals with variants in transamidase and remodelling stage genes of the GPI-AP (P = 0.046). Forty individuals had intractable epilepsy. The majority of individuals experienced delayed or absent speech (95%), motor delay with non-ambulance (64%), and severe-to-profound DD/ID (59%). Individuals with a developmental epileptic encephalopathy (51%) were at greater risk of intractable epilepsy (P = 0.003), non-ambulance (P = 0.035), ongoing enteral feeds (P < 0.001) and cortical visual impairment (P = 0.007). Serial neuroimaging showed progressive cerebral volume loss in 87.5% and progressive cerebellar atrophy in 70.8%, indicating a neurodegenerative process. Genetic analyses identified 93 unique variants (106 total), including 22 novel variants. Exploratory analyses of genotype-phenotype correlations using unsupervised hierarchical clustering identified novel genotypic predictors of clinical phenotype and long-term outcome with meaningful implications for management. In summary, we expand both the mild and severe phenotypic extremities of the IGDs, provide insights into their neurological basis, and vitally, enable meaningful genetic counselling for affected individuals and their families.
遗传性糖基磷脂酰肌醇缺乏症(IGDs)是一组罕见的多系统疾病,由糖基磷脂酰肌醇锚定途径(GPI-AP)基因的致病变异引起。尽管至少31个GPI-AP基因中的24个与人类神经遗传病相关,但先前的报告仅限于单个基因,未将GPI-AP作为一个整体考虑,且自然史数据有限。在这项多国回顾性观察研究中,我们系统分析了来自75个独特家庭的83例IGD患者的分子谱、表型特征和自然史,其中包括70例新报告的患者;这是迄今为止最大的单队列研究。核心临床特征为发育迟缓或智力残疾(DD/ID,90%)、癫痫发作(83%)、肌张力减退(72%)和运动症状((64%))。具有预后和生物学意义的神经影像学特征包括脑萎缩((75%))、小脑萎缩((60%))、胼胝体异常((57%))和中央被盖束对称受限扩散((60%))。61例患者有多系统受累,包括胃肠道((66%))、心脏((19%))和肾脏((14%))异常。虽然82%的患者有畸形特征,但没有单一畸形特征的患病率超过30%,这表明存在显著的表型异质性。所有患者均有随访数据,其中15例在撰写本文时已死亡。癫痫发作的中位年龄为6个月。GPI-AP合成阶段基因变异的患者癫痫发作开始时间显著短于GPI-AP转酰胺酶和重塑阶段基因变异的患者((P = 0.046))。40例患者患有难治性癫痫。大多数患者出现言语延迟或缺失((95%))、运动发育迟缓且无法行走((64%))以及重度至极重度DD/ID((59%))。患有发育性癫痫性脑病的患者((51%))发生难治性癫痫((P = 0.003))、无法行走((P = 0.035))、持续肠内喂养((P < 0.001))和皮质视觉障碍((P = 0.007))的风险更高。系列神经影像学检查显示,87.5%的患者脑容量逐渐减少,70.8%的患者小脑萎缩逐渐加重,提示存在神经退行性变过程。基因分析鉴定出93个独特变异(共106个),其中包括22个新变异。使用无监督层次聚类对基因型-表型相关性进行的探索性分析确定了临床表型和长期预后的新基因型预测指标,对治疗具有重要意义。总之,我们扩展了IGDs的轻度和重度表型范围,深入了解了其神经学基础,至关重要的是,为受影响的个体及其家庭提供了有意义的遗传咨询。