Hubei Key Laboratory of Biological Resources Protection and Utilization, Enshi, China, Enshi.
Hubei Key Laboratory of Selenium Resource Research and Biological Application, Enshi, China, 44500.
BMC Plant Biol. 2024 Mar 19;24(1):199. doi: 10.1186/s12870-024-04898-9.
The selenomethionine cycle (SeMTC) is a crucial pathway for the metabolism of selenium. The basic bioinformatics and functions of four enzymes involved in the cycle including S-adenosyl-methionine synthase (MAT), SAM-dependent methyltransferase (MTase), S-adenosyl-homocysteine hydrolase (SAHH) and methionine synthase (MTR), have been extensively reported in many eukaryotes. The identification and functional analyses of SeMTC genes/proteins in Cardamine hupingshanensis and their response to selenium stress have not yet been reported.
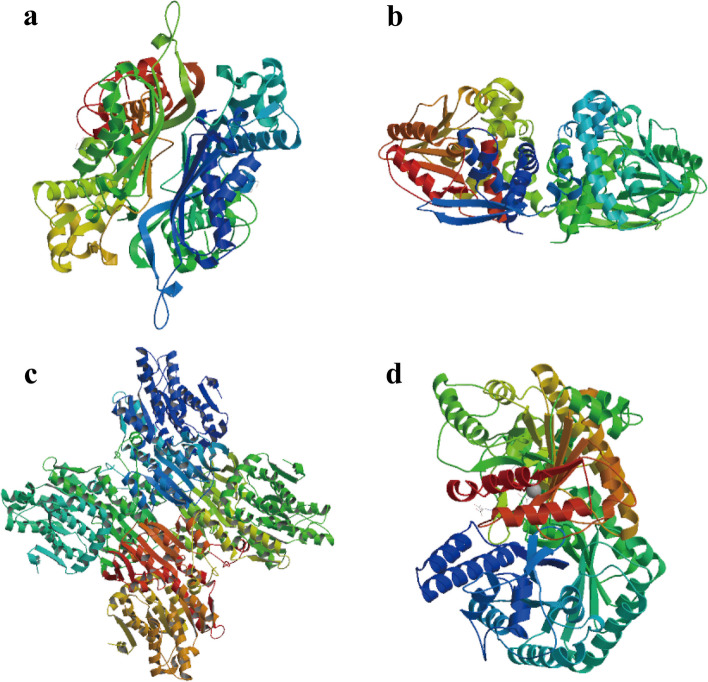
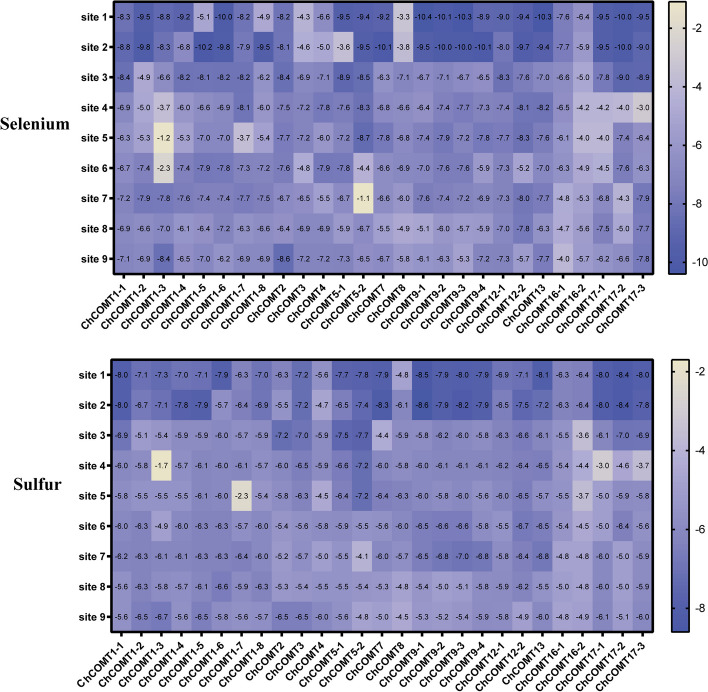
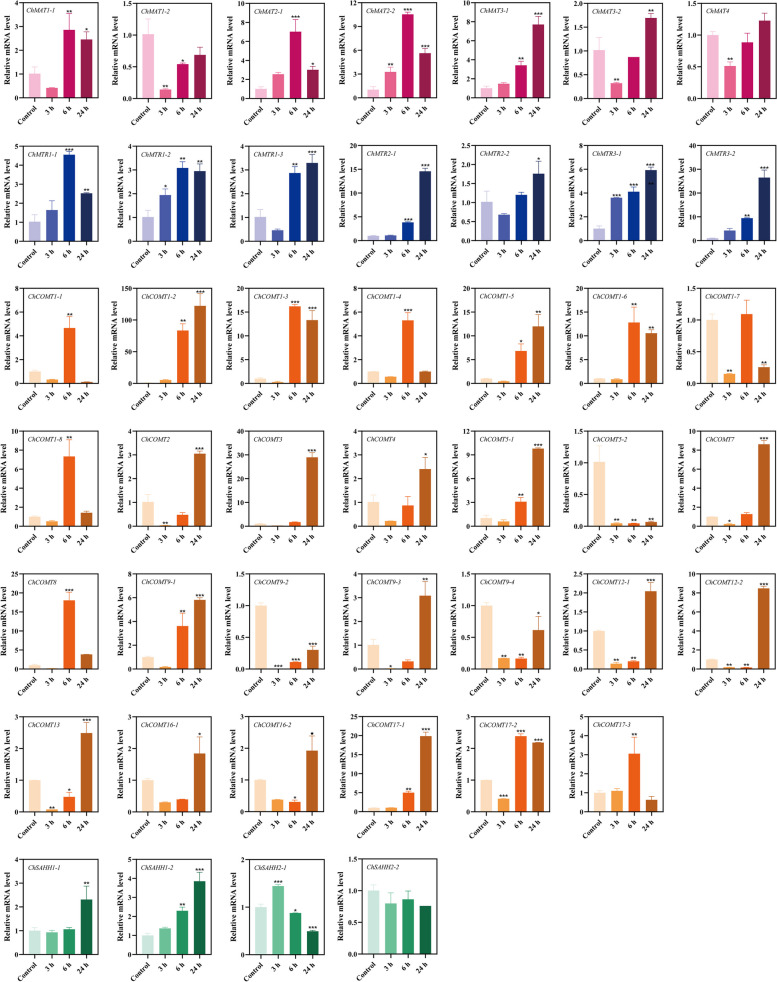
In this study, 45 genes involved in SeMTC were identified in the C. hupingshanensis genome. Phylogenetic analysis showed that seven genes from ChMAT were clustered into four branches, twenty-seven genes from ChCOMT were clustered into two branches, four genes from ChSAHH were clustered into two branches, and seven genes from ChMTR were clustered into three branches. These genes were resided on 16 chromosomes. Gene structure and homologous protein modeling analysis illustrated that proteins in the same family are relatively conserved and have similar functions. Molecular docking showed that the affinity of SeMTC enzymes for selenium metabolites was higher than that for sulfur metabolites. The key active site residues identified for ChMAT were Ala and Lys, while Leu and Gly were determined as the crucial residues for ChCOMT. For ChSAHH, the essential active site residues were found to be Asn, Asp and Thr. Ile, Ser, Asp, and His were identified as the critical active site residues for ChMTR. In addition, the results of the expression levels of four enzymes under selenium stress revealed that ChMAT3-1 genes were upregulated approximately 18-fold, ChCOMT9-1 was upregulated approximately 38.7-fold, ChSAHH1-2 was upregulated approximately 11.6-fold, and ChMTR3-2 genes were upregulated approximately 28-fold. These verified that SeMTC enzymes were involved in response to selenium stress to varying degrees.
The results of this research are instrumental for further functional investigation of SeMTC in C. hupingshanensis. This also lays a solid foundation for deeper investigations into the physiological and biochemical mechanisms underlying selenium metabolism in plants.
硒代蛋氨酸循环(SeMTC)是硒代谢的关键途径。在许多真核生物中,已广泛报道了参与该循环的四种酶,包括 S-腺苷甲硫氨酸合成酶(MAT)、SAM 依赖性甲基转移酶(MTase)、S-腺苷同型半胱氨酸水解酶(SAHH)和蛋氨酸合成酶(MTR)的基本生物信息学和功能。然而,在山黧豆中,尚未报道 SeMTC 基因/蛋白的鉴定和功能分析及其对硒胁迫的响应。
本研究在山黧豆基因组中鉴定出 45 个参与 SeMTC 的基因。系统发育分析表明,来自 ChMAT 的 7 个基因聚为 4 个分支,来自 ChCOMT 的 27 个基因聚为 2 个分支,来自 ChSAHH 的 4 个基因聚为 2 个分支,来自 ChMTR 的 7 个基因聚为 3 个分支。这些基因位于 16 条染色体上。基因结构和同源蛋白建模分析表明,同一家族的蛋白质相对保守,具有相似的功能。分子对接表明,SeMTC 酶对硒代谢物的亲和力高于对硫代谢物的亲和力。确定 ChMAT 的关键活性位点残基为 Ala 和 Lys,而 Leu 和 Gly 被确定为 ChCOMT 的关键残基。对于 ChSAHH,关键的活性位点残基被鉴定为 Asn、Asp 和 Thr。对于 ChMTR,确定 Ile、Ser、Asp 和 His 为关键活性位点残基。此外,硒胁迫下四种酶表达水平的结果表明,ChMAT3-1 基因上调约 18 倍,ChCOMT9-1 上调约 38.7 倍,ChSAHH1-2 上调约 11.6 倍,ChMTR3-2 基因上调约 28 倍。这些结果证实,SeMTC 酶不同程度地参与了对硒胁迫的响应。
本研究结果为进一步研究山黧豆中 SeMTC 的功能提供了依据,也为深入研究植物硒代谢的生理生化机制奠定了基础。