Palova Hana, Das Anirban, Pokorna Petra, Bajciova Viera, Pavelka Zdenek, Jezova Marta, Pal Karol, Dimayacyac Jose R, Negm Logine, Stengs Lucie, Bianchi Vanessa, Vejmelkova Klara, Noskova Kristyna, Jarosova Marie, Mejstrikova Sona, Mudry Peter, Kyr Michal, Merta Tomas, Tinka Pavel, Drabova Klara, Aulicka Stefania, Jugas Robin, Tabori Uri, Slaby Ondrej, Sterba Jaroslav
Central European Institute of Technology, Masaryk University, Brno, Czech Republic.
Department of Biology, Faculty of Medicine, Masaryk University, Brno, Czech Republic.
NPJ Precis Oncol. 2024 May 21;8(1):110. doi: 10.1038/s41698-024-00597-8.
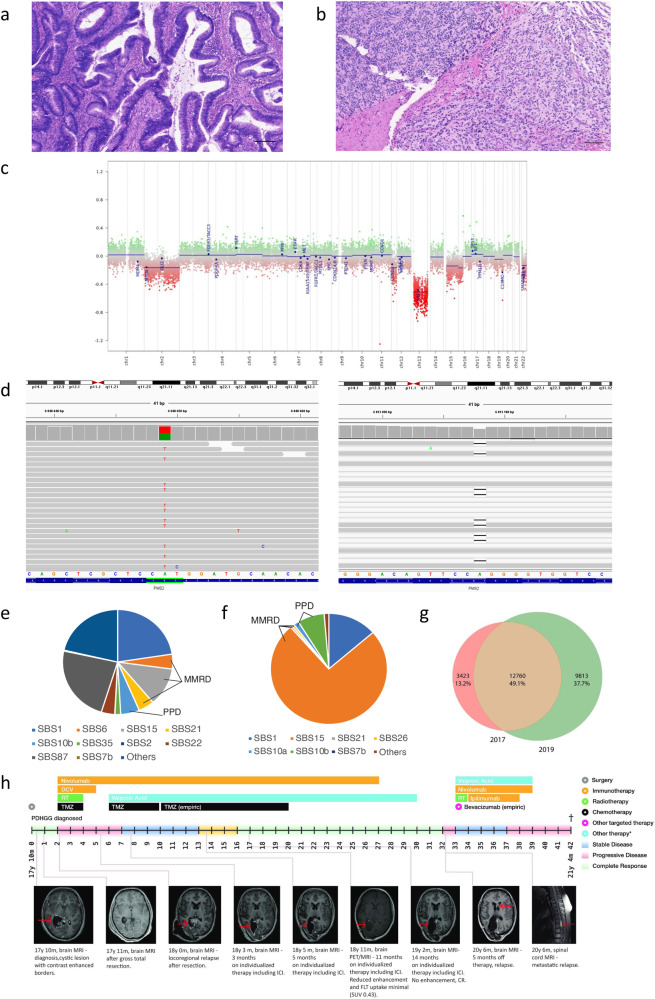
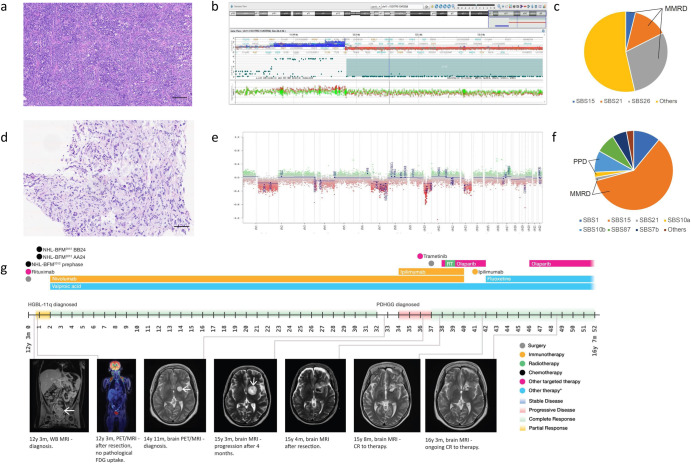
Constitutional mismatch repair deficiency (CMMRD) is a rare syndrome characterized by an increased incidence of cancer. It is caused by biallelic germline mutations in one of the four mismatch repair genes (MMR) genes: MLH1, MSH2, MSH6, or PMS2. Accurate diagnosis accompanied by a proper molecular genetic examination plays a crucial role in cancer management and also has implications for other family members. In this report, we share the impact of the diagnosis and challenges during the clinical management of two brothers with CMMRD from a non-consanguineous family harbouring compound heterozygous variants in the PMS2 gene. Both brothers presented with different phenotypic manifestations and cancer spectrum. Treatment involving immune checkpoint inhibitors significantly contributed to prolonged survival in both patients affected by lethal gliomas. The uniform hypermutation also allowed immune-directed treatment using nivolumab for the B-cell lymphoma, thereby limiting the intensive chemotherapy exposure in this young patient who remains at risk for subsequent malignancies.
先天性错配修复缺陷(CMMRD)是一种罕见综合征,其特征为癌症发病率增加。它由四个错配修复(MMR)基因之一的双等位基因种系突变引起:MLH1、MSH2、MSH6或PMS2。准确诊断并辅以适当的分子遗传学检查在癌症管理中起着关键作用,对其他家庭成员也有影响。在本报告中,我们分享了对来自一个非近亲家庭的两名患有CMMRD的兄弟进行临床管理时诊断的影响和挑战,该家庭的PMS2基因存在复合杂合变异。两兄弟表现出不同的表型表现和癌症谱。涉及免疫检查点抑制剂的治疗显著延长了两名患有致命性胶质瘤患者的生存期。一致的高度突变也使得可以使用纳武单抗对B细胞淋巴瘤进行免疫导向治疗,从而减少了这位仍有后续恶性肿瘤风险的年轻患者的强化化疗暴露。