Parvin Shanaz, Dey Anita Rani, Shohana Nusrat Nowrin, Talukder Md Hasanuzzaman, Alam Mohammad Zahangir
Department of Parasitology, Faculty of Veterinary Science, Bangladesh Agricultural University, Mymensingh 2202, Bangladesh.
Department of Para-Clinical Courses, Faculty of Veterinary and Animal Sciences, Gono Bishwabidyalay, Dhaka, Bangladesh.
Saudi J Biol Sci. 2024 Aug;31(8):104030. doi: 10.1016/j.sjbs.2024.104030. Epub 2024 May 26.
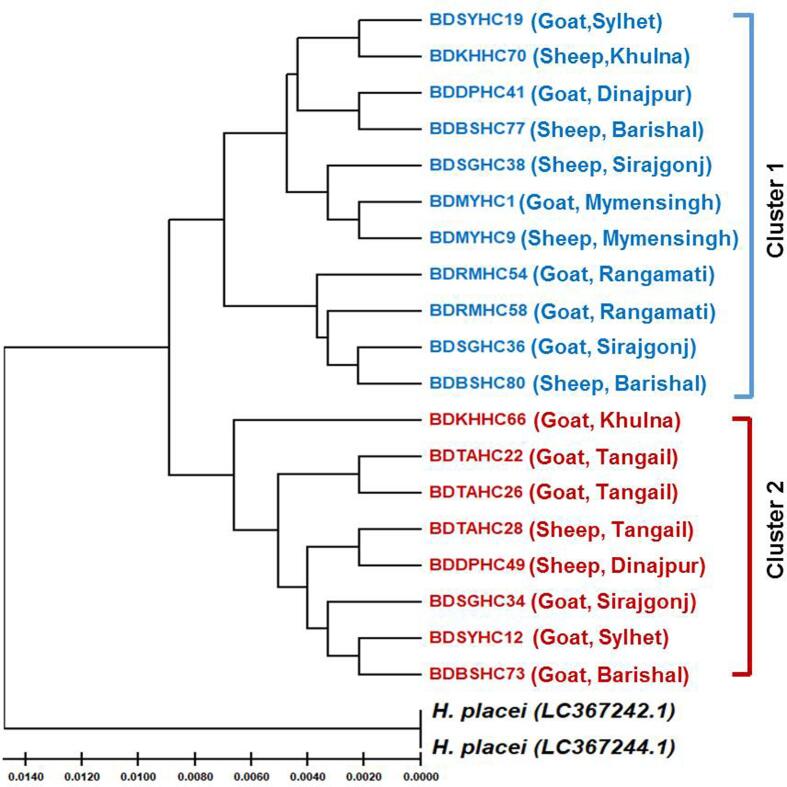
a stomach worm, is prevalent in ruminants worldwide. They particularly hamper profitable small ruminant production. Here, we estimate the genetic variation of collected from slaughtered goats and sheep from various geographic zones of Bangladesh using multiple genes. To perform this, adult parasites were isolated from the abomasum of slaughtered animals (sheep and goats). Among them, 79 male were identified by microscopy. Following the extraction of DNA, and genes were amplified and subsequently considered for sequencing. After alignment and editing, sequences were analyzed to find out sequence variation, diversity pattern of genes, and population genetics of isolates. Among the sequence data, the analyses identified 19 genotypes of and 77 haplotypes of genes. The diversity of nucleotides was 0.0103 for and 0.029 for gene. The dendogram constructed by the genotype and haplotype sequences of revealed that two populations were circulating in Bangladesh without any demarcation of host and geographic regions. Analysis of population genetics demonstrated a high flow of genes (89.2 %) within the population of the worm in Bangladesh. The Fst value showed very little amount of genetic difference among the worm populations of Bangladesh but marked genetic variation between different continents. The findings are expected to help explain the risks of anthelmintic resistance and the transmission pattern of the parasite, and also provide a control strategy against .
一种胃虫在全球反刍动物中普遍存在。它们尤其阻碍了盈利性小反刍动物的生产。在此,我们使用多个基因估计了从孟加拉国不同地理区域屠宰的山羊和绵羊身上采集的[寄生虫名称未给出]的遗传变异。为此,从屠宰动物(绵羊和山羊)的皱胃中分离出成年寄生虫。其中,通过显微镜鉴定出79只雄性[寄生虫名称未给出]。提取DNA后,[基因名称未给出]和[基因名称未给出]基因被扩增,随后进行测序。经过比对和编辑后,对序列进行分析以找出序列变异、基因多样性模式以及分离株的群体遗传学特征。在序列数据中,分析确定了[寄生虫名称未给出]的19种基因型和[基因名称未给出]基因的77种单倍型。[寄生虫名称未给出]基因的核苷酸多样性为0.0103,[基因名称未给出]基因的核苷酸多样性为0.029。由[寄生虫名称未给出]的基因型和单倍型序列构建的树形图显示,在孟加拉国存在两个种群在循环,且没有宿主和地理区域的划分。群体遗传学分析表明,在孟加拉国该蠕虫种群内基因流动率很高(89.2%)。Fst值显示孟加拉国蠕虫种群之间的遗传差异非常小,但不同大陆之间存在明显的遗传变异。这些发现有望有助于解释抗驱虫药耐药性的风险和寄生虫的传播模式,也为针对[寄生虫名称未给出]的控制策略提供依据。