From the Department of Neurology (L.L., C.L., M.C.-Á., Á.C., A.V., L.Q., E.G., M.O.), Neuromuscular Diseases Unit; Department of Genetics (A.S.-C., B.R.-S.), Hospital de la Santa Creu i Sant Pau, Universitat Autònoma de Barcelona; Department of Neurology (C.D.-G.), Neuromuscular Diseases Unit, Hospital Universitario 12 de Octubre. Research Institute imas12, Biomedical Network Research Centre on Rare Diseases (CIBERER), Instituto de Salud Carlos III, Madrid, Spain; Université Paris-Est Créteil (E.M.), INSERM, U955 IMRB; AP-HP, Hôpital Mondor, FHU SENEC, Service d'Histologie, Créteil, France; Department of Neurology (S.K.), Neuromuscular Diseases Unit, Osakidetza Basque Health Service, Basurto University Hospital, Universidad del País Vasco, Bilbao; Institut de Recerca Sant Pau (IR Sant Pau) (B.R.-S., R.B., C.L., L.Q., E.G., M.O.), Barcelona; Biomedical Network Research Centre on Rare Diseases (CIBERER), Madrid; Genomic Instability Syndromes and DNA Repair Group and Join Research Unit on Genomic Medicine UAB (B.R.-S.), Institut de Recerca Sant Pau (IR Sant Pau), Hospital de la Santa Creu i Sant Pau; Immunology Department (O.C., A.M.), Hospital de la Santa Creu i Sant Pau, Universitat Autònoma de Barcelona, Institut de Recerca Sant Pau (IR Sant Pau), Barcelona, Spain; Department of Genetics (A.D.), Craiova University Hospital, Romania; Neuropaediatrics Department (A.N.O.), Neuromuscular Diseases Unit, Hospital Sant Joan de Déu, Fundación Sant Joan de Déu, CIBERER - ISC III; Neurology Department (A.P., L.G.-M.), Neuromuscular Unit, IDIBELL-Hospital de Bellvitge, Hospitalet de Llobregat, Barcelona; Pathology Department (A.H.-L.), Neuropathology Unit, Hospital Universitario 12 de Octubre, Madrid; Pathology Department (C.J.), Institut Pediàtric de Recerca, Hospital Sant Joan de Déu, and MetabERN, Barcelona; Biomedical Network Research Centre on Rare Diseases (CIBERER), Instituto de Salud Carlos III, Madrid; Department of Neurology (L.G.-M.), Hospital de Viladecans, Barcelona; and Department of Genetics (A.A.), Hospital Universitario 12 de Octubre, Research Institute imas12, Madrid, Spain.
Neurol Neuroimmunol Neuroinflamm. 2024 Sep;11(5):e200285. doi: 10.1212/NXI.0000000000200285. Epub 2024 Aug 6.
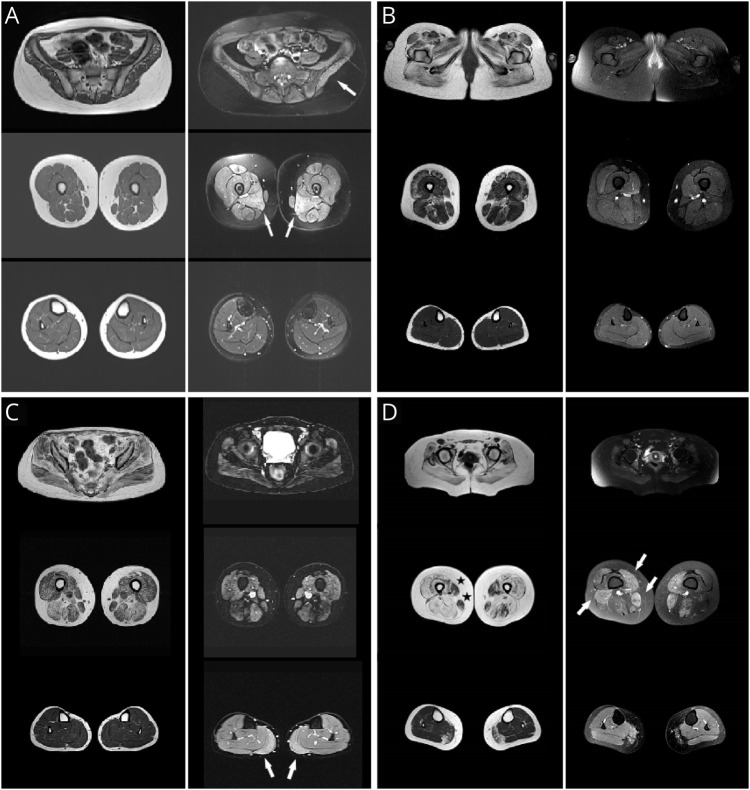
Immune-mediated necrotizing myopathy (IMNM) caused by antibodies against 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) is an inflammatory myopathy that has been epidemiologically correlated with previous statin exposure. We characterized in detail a series of 11 young statin-naïve patients experiencing a chronic disease course mimicking a limb-girdle muscular dystrophy. With the hypothesis that HMGCR upregulation may increase immunogenicity and trigger the production of autoantibodies, our aim was to expand pathophysiologic knowledge of this distinct phenotype.
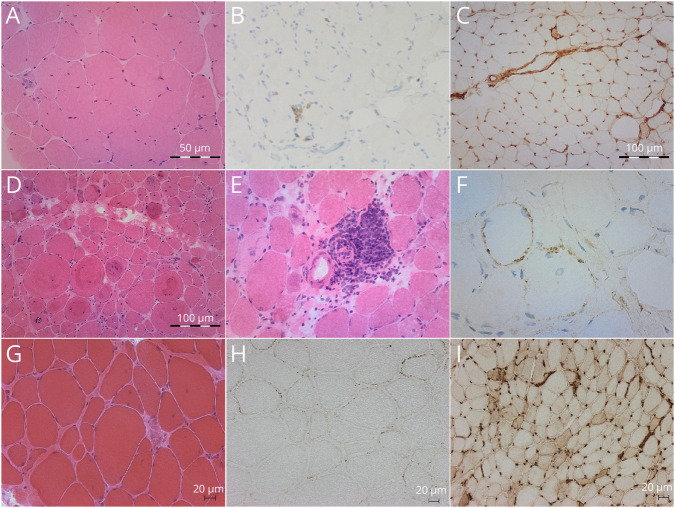
Clinical and epidemiologic data, autoantibody titers, creatine kinase (CK) levels, response to treatment, muscle imaging, and muscle biopsies were assessed. HMGCR expression in patients' muscle was assessed by incubating sections of affected patients with purified anti-HMGCR+ serum. Whole-exome sequencing (WES) with a special focus on cholesterol biosynthesis-related genes and high-resolution human leukocyte antigen (HLA) typing were performed.
Patients, aged 3-25 years and mostly female (90.9%), presented with subacute proximal weakness progressing over many years and high CK levels (>1,000 U/L). Diagnostic delay ranged from 3 to 27 years. WES did not reveal any pathogenic variants. HLA-DRB1*11:01 carrier frequency was 60%, a significantly higher proportion than in the control population. No upregulation or mislocalization of the enzyme in statin-exposed or statin-naïve anti-HMGCR+ patients was observed, compared with controls.
WES of a cohort of patients with dystrophy-like anti-HMGCR IMNM did not reveal any common rare variants of any gene, including cholesterol biosynthesis-related genes. HLA analysis showed a strong association with HLA-DRB1*11:01, previously mostly described in statin-exposed adult patients; consequently, a common immunogenic predisposition should be suspected, irrespective of statin exposure. Moreover, we were unable to conclusively demonstrate muscle upregulation/mislocalization of HMGCR in IMNM, whether or not driven by statins.
由 3-羟基-3-甲基戊二酰辅酶 A 还原酶(HMGCR)抗体引起的免疫介导的坏死性肌病(IMNM)是一种炎症性肌病,已在流行病学上与先前的他汀类药物暴露相关。我们详细描述了一系列 11 例年轻的他汀类药物初治患者,他们经历了一种类似于肢带型肌营养不良的慢性疾病过程。我们假设 HMGCR 的上调可能会增加免疫原性并触发自身抗体的产生,因此我们的目的是扩展这种独特表型的病理生理学知识。
评估了临床和流行病学数据、自身抗体滴度、肌酸激酶(CK)水平、治疗反应、肌肉成像和肌肉活检。通过将受影响患者的切片与纯化的抗-HMGCR+血清孵育,评估患者肌肉中的 HMGCR 表达。进行全外显子组测序(WES),特别关注胆固醇生物合成相关基因和高分辨率人类白细胞抗原(HLA)分型。
患者年龄 3-25 岁,大多数为女性(90.9%),表现为亚急性近端肌无力,病程多年,CK 水平升高(>1,000 U/L)。诊断延迟时间为 3-27 年。WES 未发现任何致病性变异。HLA-DRB1*11:01 携带者频率为 60%,明显高于对照组。与对照组相比,在他汀类药物暴露或他汀类药物初治抗-HMGCR+患者中,均未观察到该酶的上调或定位错误。
对一组具有肌病样抗-HMGCR IMNM 的患者进行 WES 未发现任何基因的常见罕见变异,包括胆固醇生物合成相关基因。HLA 分析显示与 HLA-DRB1*11:01 强烈相关,以前主要描述于他汀类药物暴露的成年患者;因此,应怀疑存在共同的免疫易感性,而不论是否存在他汀类药物暴露。此外,我们无法明确证明无论是否由他汀类药物驱动,IMNM 中 HMGCR 的肌肉上调/定位错误。