Institute of Metabolism and Systems Research, University of Birmingham, Birmingham, United Kingdom.
Centre for Endocrinology, Diabetes and Metabolism, University Hospitals Birmingham NHS Foundation Trust, Birmingham Health Partners, Birmingham, United Kingdom.
Eur J Endocrinol. 2024 Aug 30;191(3):334-344. doi: 10.1093/ejendo/lvae106.
Primary bilateral macronodular adrenal hyperplasia (PBMAH) is a rare cause of Cushing's syndrome. Individuals with PBMAH and glucose-dependent insulinotropic polypeptide (GIP)-dependent Cushing's syndrome due to ectopic expression of the GIP receptor (GIPR) typically harbor inactivating KDM1A sequence variants. Primary unilateral macronodular adrenal hyperplasia (PUMAH) with concomitant glucocorticoid and androgen excess has never been encountered or studied.
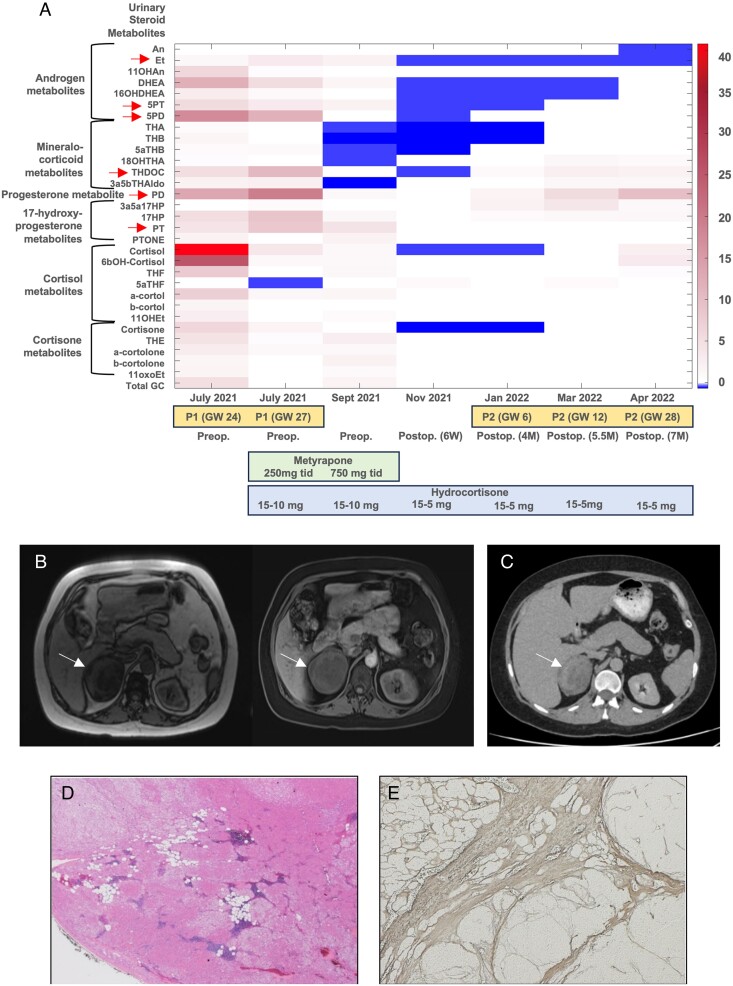
We investigated a woman with a large, heterogeneous adrenal mass and severe adrenocorticotropic hormone-independent glucocorticoid and androgen excess, a biochemical presentation typically suggestive of adrenocortical carcinoma. The patient presented during pregnancy (22nd week of gestation) and reported an 18-month history of oligomenorrhea, hirsutism, and weight gain. We undertook an exploratory study with detailed histopathological and genetic analysis of the resected adrenal mass and leukocyte DNA collected from the patient and her parents.
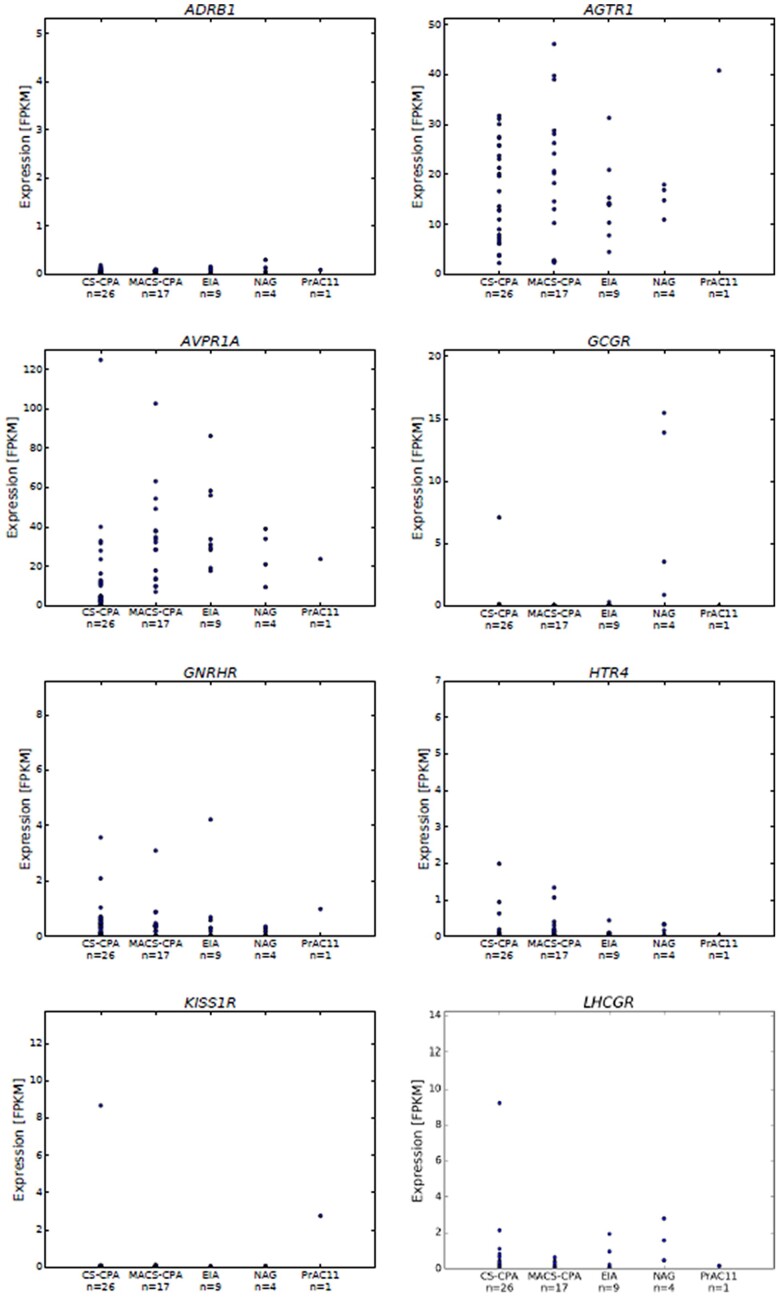
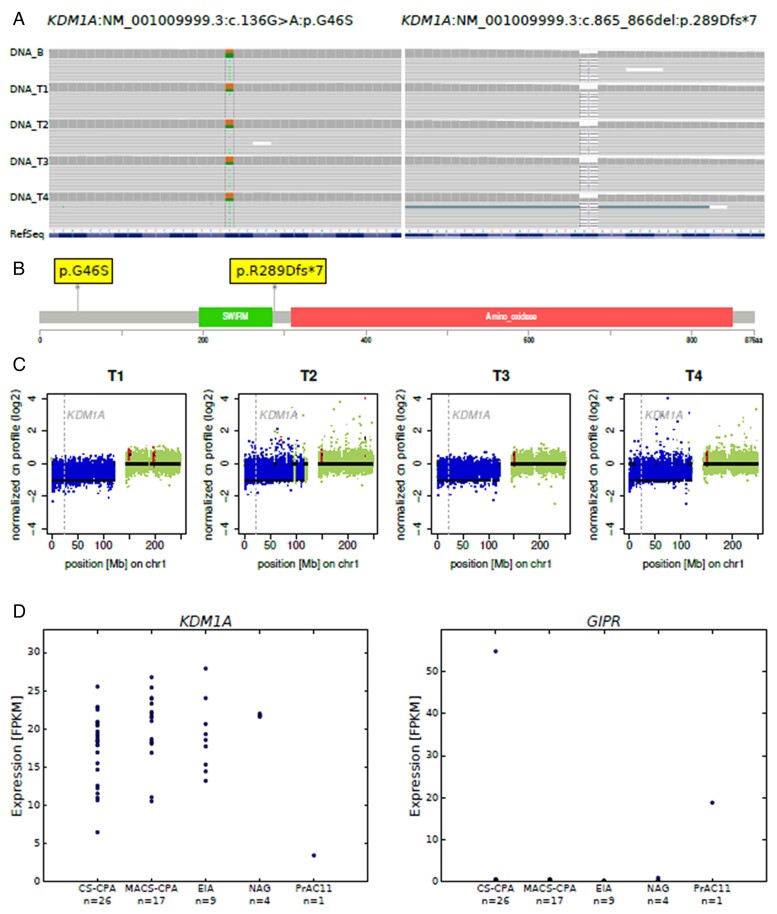
Histopathology revealed benign macronodular adrenal hyperplasia. Imaging showed a persistently normal contralateral adrenal gland. Whole-exome sequencing of 4 representative nodules detected KDM1A germline variants, benign NM_001009999.3:c.136G > A:p.G46S, and likely pathogenic NM_001009999.3:exon6:c.865_866del:p.R289Dfs7. Copy number variation analysis demonstrated an additional somatic loss of the KDM1A wild-type allele on chromosome 1p36.12 in all nodules. RNA sequencing of a representative nodule showed low/absent KDM1A expression and increased GIPR expression compared with 52 unilateral sporadic adenomas and 4 normal adrenal glands. Luteinizing hormone/chorionic gonadotropin receptor expression was normal. Sanger sequencing confirmed heterozygous KDM1A variants in both parents (father: p.R289Dfs7 and mother: p.G46S) who showed no clinical features suggestive of glucocorticoid or androgen excess.
We investigated the first PUMAH associated with severe Cushing's syndrome and concomitant androgen excess, suggesting pathogenic mechanisms involving KDM1A.
原发性双侧大结节性肾上腺增生症(PBMAH)是库欣综合征的罕见病因。由于葡萄糖依赖性胰岛素促分泌多肽受体(GIPR)的异位表达而患有 PBMAH 和 GIP 依赖性库欣综合征的个体通常携带失活的 KDM1A 序列变异。伴有糖皮质激素和雄激素过度分泌的原发性单侧大结节性肾上腺增生症(PUMAH)从未遇到或研究过。
我们研究了一位患有大型异质性肾上腺肿块和严重促肾上腺皮质激素非依赖性糖皮质激素和雄激素过度分泌的女性患者,这种生化表现通常提示为肾上腺皮质癌。该患者在妊娠(22 周妊娠)期间就诊,并报告了 18 个月的月经稀少、多毛症和体重增加病史。我们对切除的肾上腺肿块和从患者及其父母采集的白细胞 DNA 进行了详细的组织病理学和遗传学研究。
组织病理学显示为良性大结节性肾上腺增生。影像学显示对侧肾上腺持续正常。对 4 个代表性结节进行全外显子组测序检测到 KDM1A 种系变异,良性 NM_001009999.3:c.136G > A:p.G46S 和可能致病性 NM_001009999.3:exon6:c.865_866del:p.R289Dfs7。拷贝数变异分析表明,所有结节中 1p36.12 染色体上 KDM1A 野生型等位基因的额外体细胞丢失。代表性结节的 RNA 测序显示与 52 例单侧散发性腺瘤和 4 例正常肾上腺相比,KDM1A 表达降低/缺失,GIPR 表达增加。黄体生成素/绒毛膜促性腺激素受体表达正常。Sanger 测序证实父母双方均存在杂合性 KDM1A 变异(父亲:p.R289Dfs7,母亲:p.G46S),他们均无糖皮质激素或雄激素过度分泌的临床特征。
我们研究了首例与严重库欣综合征和伴发雄激素过度分泌相关的 PUMAH,提示涉及 KDM1A 的致病机制。