Drug Theoretics and Cheminformatics Laboratory, Department of Pharmaceutical Technology, Jadavpur University, Kolkata, 700 032, India.
Sci Rep. 2024 Sep 6;14(1):20812. doi: 10.1038/s41598-024-71892-4.
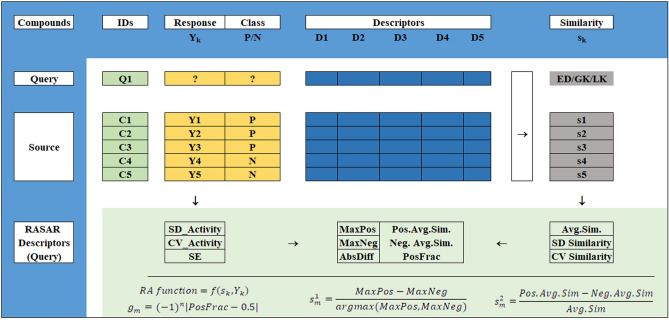
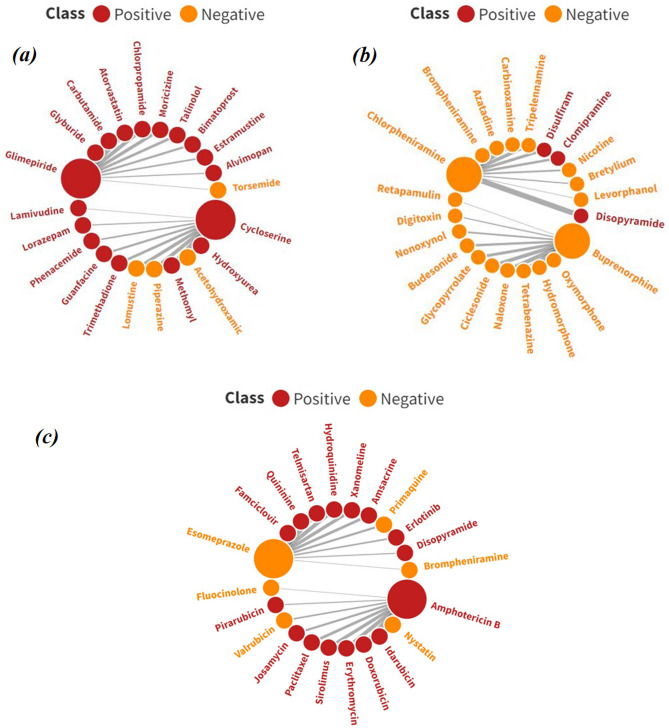
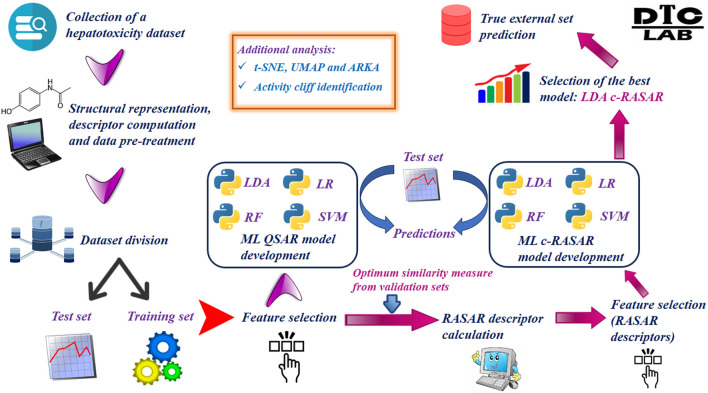
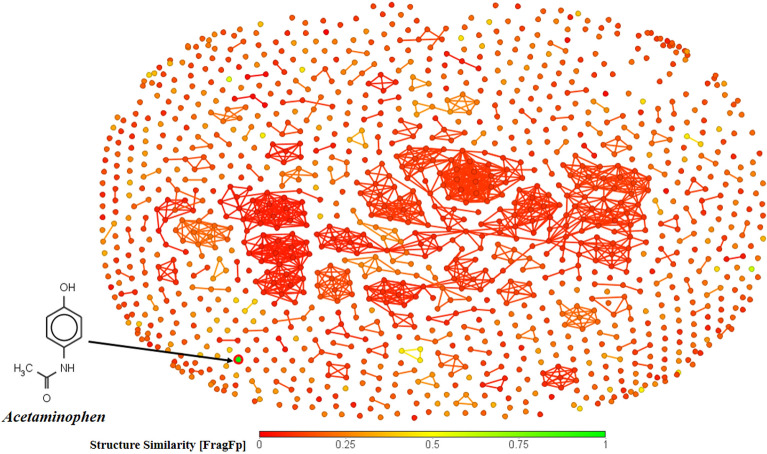
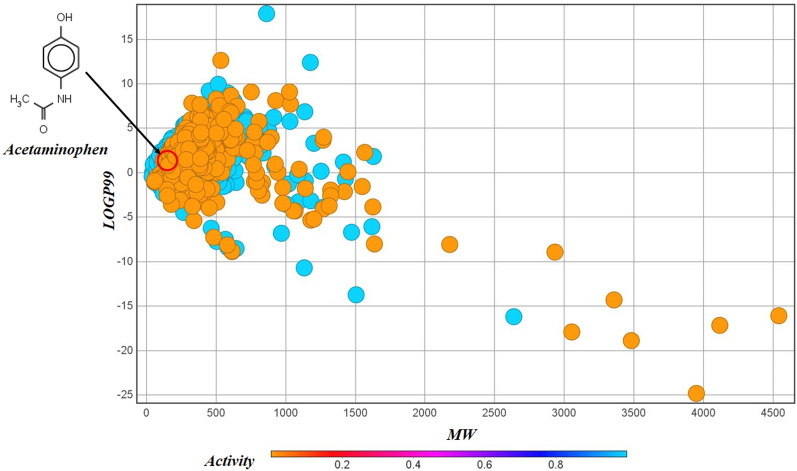
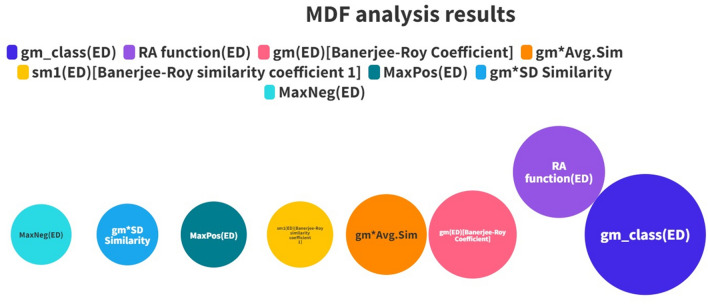
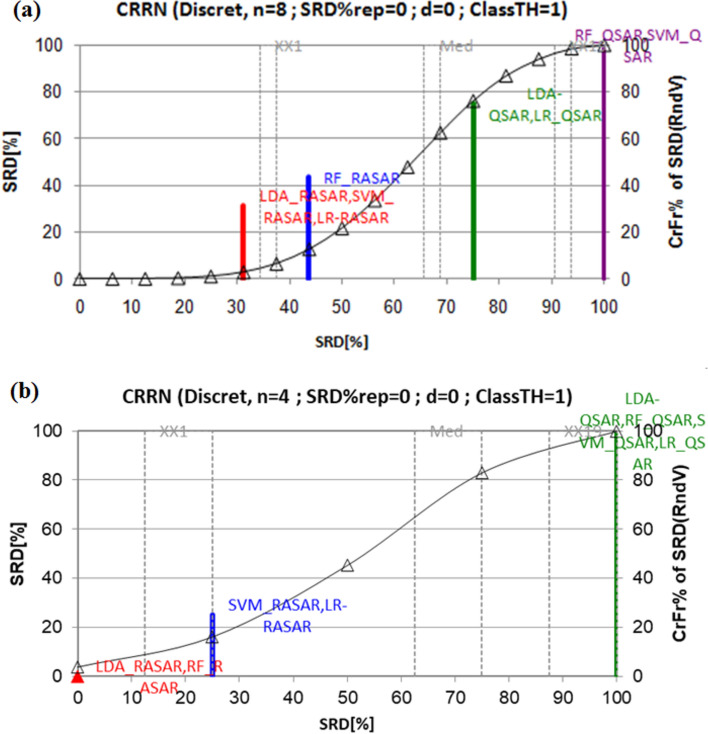
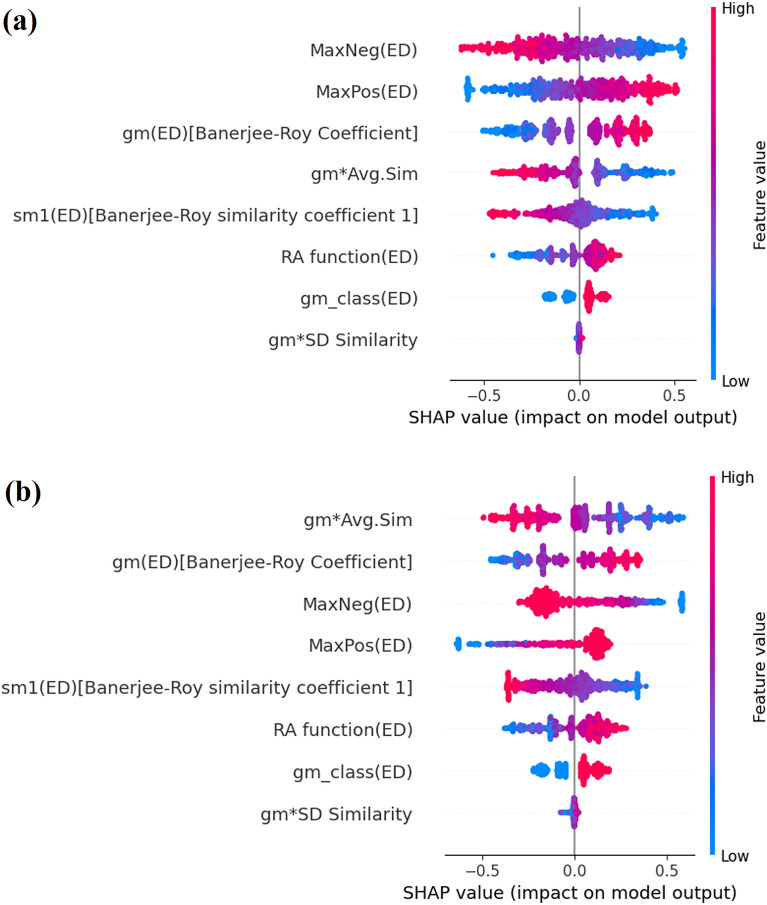
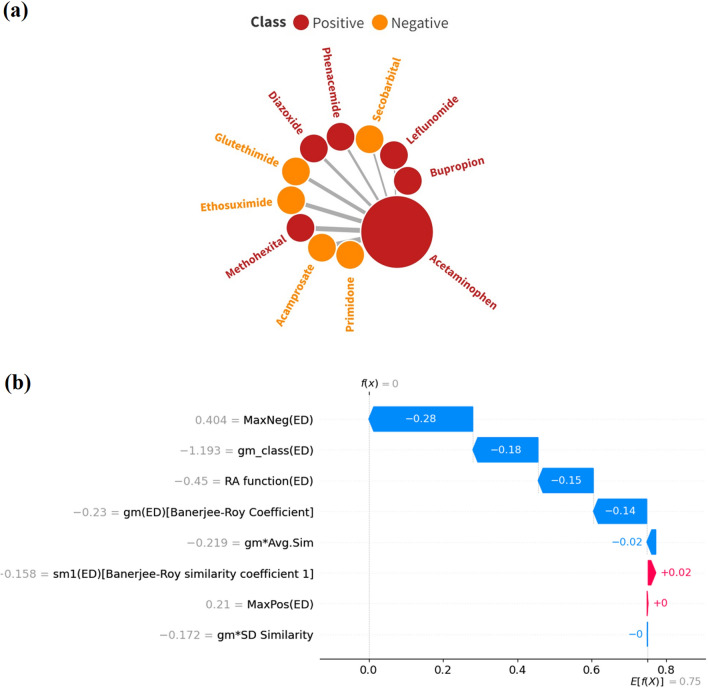
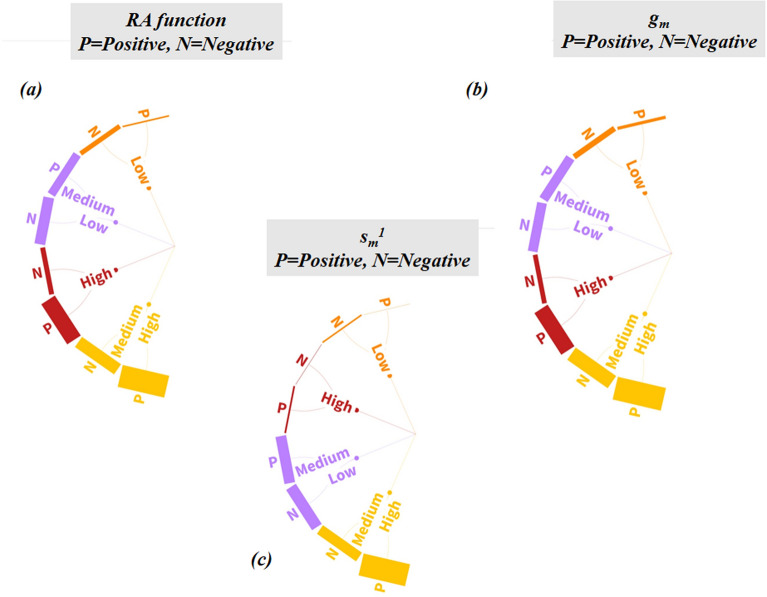
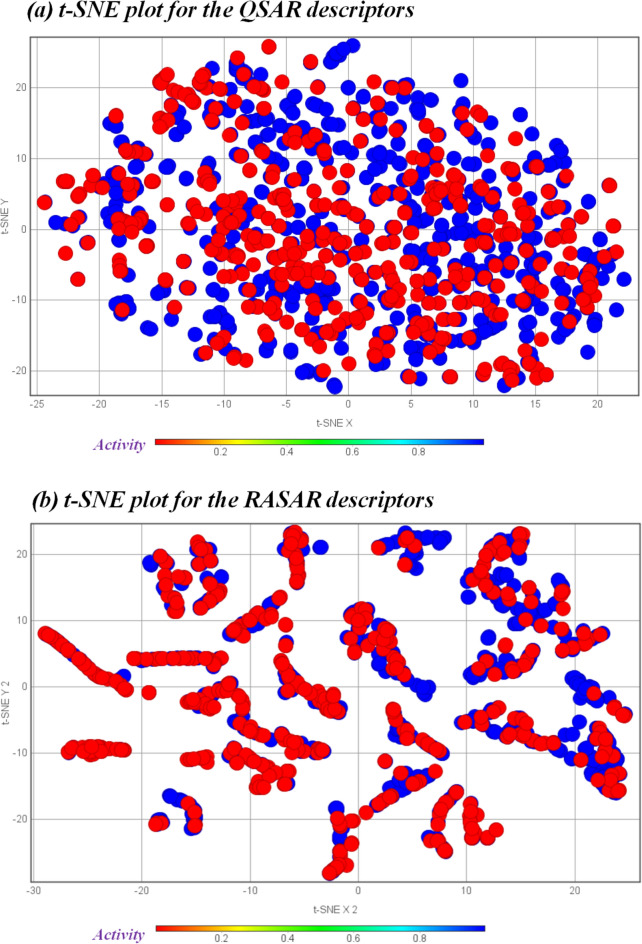
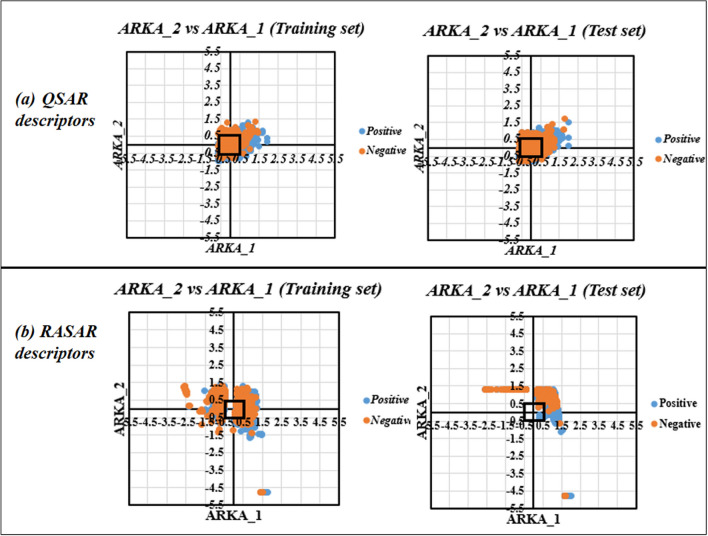
With the exponential progress in the field of cheminformatics, the conventional modeling approaches have so far been to employ supervised and unsupervised machine learning (ML) and deep learning models, utilizing the standard molecular descriptors, which represent the structural, physicochemical, and electronic properties of a particular compound. Deviating from the conventional approach, in this investigation, we have employed the classification Read-Across Structure-Activity Relationship (c-RASAR), which involves the amalgamation of the concepts of classification-based quantitative structure-activity relationship (QSAR) and Read-Across to incorporate Read-Across-derived similarity and error-based descriptors into a statistical and machine learning modeling framework. ML models developed from these RASAR descriptors use similarity-based information from the close source neighbors of a particular query compound. We have employed different classification modeling algorithms on the selected QSAR and RASAR descriptors to develop predictive models for efficient prediction of query compounds' hepatotoxicity. The predictivity of each of these models was evaluated on a large number of test set compounds. The best-performing model was also used to screen a true external data set. The concepts of explainable AI (XAI) coupled with Read-Across were used to interpret the contributions of the RASAR descriptors in the best c-RASAR model and to explain the chemical diversity in the dataset. The application of various unsupervised dimensionality reduction techniques like t-SNE and UMAP and the supervised ARKA framework showed the usefulness of the RASAR descriptors over the selected QSAR descriptors in their ability to group similar compounds, enhancing the modelability of the dataset and efficiently identifying activity cliffs. Furthermore, the activity cliffs were also identified from Read-Across by observing the nature of compounds constituting the nearest neighbors for a particular query compound. On comparing our simple linear c-RASAR model with the previously reported models developed using the same dataset derived from the US FDA Orange Book ( https://www.accessdata.fda.gov/scripts/cder/ob/index.cfm ), it was observed that our model is simple, reproducible, transferable, and highly predictive. The performance of the LDA c-RASAR model on the true external set supersedes that of the previously reported work. Therefore, the present simple LDA c-RASAR model can efficiently be used to predict the hepatotoxicity of query chemicals.
随着化学信息学领域的指数级发展,传统的建模方法迄今为止一直是利用有监督和无监督机器学习 (ML) 和深度学习模型,利用代表特定化合物结构、物理化学和电子特性的标准分子描述符。在这项研究中,我们采用了分类读跨结构-活性关系 (c-RASAR),这与传统方法不同,它涉及到将基于分类的定量结构-活性关系 (QSAR) 和读跨的概念结合起来,将读跨衍生的相似性和基于错误的描述符纳入统计和机器学习建模框架中。从这些 RASAR 描述符中开发的 ML 模型使用特定查询化合物的近源邻居的基于相似性的信息。我们在选定的 QSAR 和 RASAR 描述符上使用了不同的分类建模算法,以开发用于有效预测查询化合物肝毒性的预测模型。这些模型中的每一个都在大量测试集化合物上进行了预测性评估。表现最好的模型也被用于筛选一个真正的外部数据集。结合了可解释人工智能 (XAI) 的概念和读跨被用于解释最佳 c-RASAR 模型中 RASAR 描述符的贡献,并解释数据集的化学多样性。应用各种无监督降维技术,如 t-SNE 和 UMAP 以及监督的 ARKA 框架,显示了 RASAR 描述符在其将相似化合物分组的能力、增强数据集的可建模性和有效地识别活性悬崖方面优于选定的 QSAR 描述符的有用性。此外,通过观察构成特定查询化合物最近邻的化合物的性质,还可以从读跨中识别活性悬崖。将我们简单的线性 c-RASAR 模型与之前使用相同数据集(源自美国 FDA 橙皮书 ( https://www.accessdata.fda.gov/scripts/cder/ob/index.cfm ))开发的报告模型进行比较,结果表明我们的模型简单、可重复、可转移且具有高度预测性。LDA c-RASAR 模型在真实外部数据集上的性能优于之前的报告工作。因此,目前简单的 LDA c-RASAR 模型可以有效地用于预测查询化学品的肝毒性。