Rauf Hasaan S, Liu Yu-Sheng, Arslan Muhammad, Solanki Surya Pratap S, Deydier Eric, Poli Rinaldo, Grabow Lars C, Harth Eva
Department of Chemistry, Center of Excellence in Polymer Chemistry (CPEC), University of Houston, 3589 Cullen Boulevard, Houston, Texas 77004, United States.
Department of Chemical and Biomolecular Engineering, University of Houston, 4726 Calhoun Rd, S222 Engineering Building 1, Houston, Texas 77204, United States.
ACS Catal. 2024 Aug 16;14(17):13136-13147. doi: 10.1021/acscatal.4c02708. eCollection 2024 Sep 6.
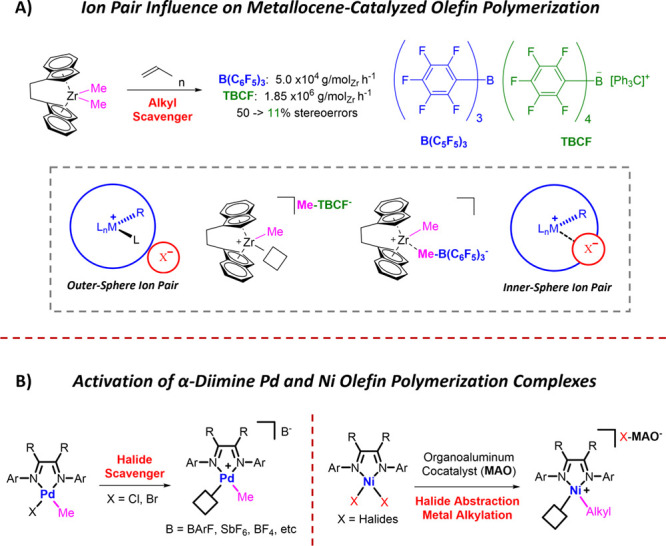
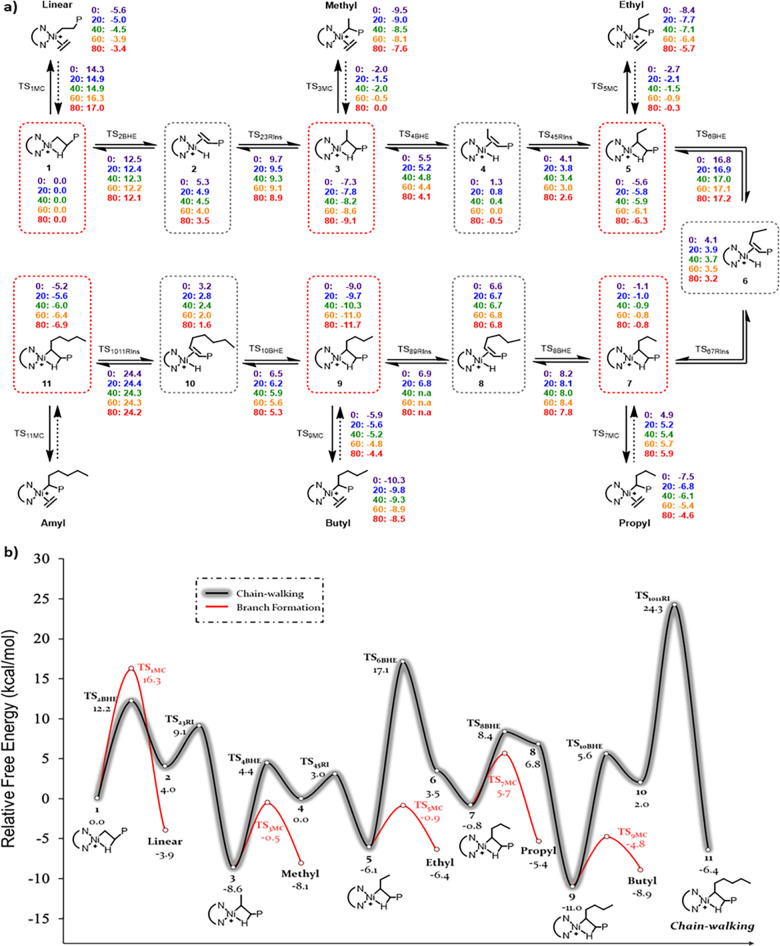
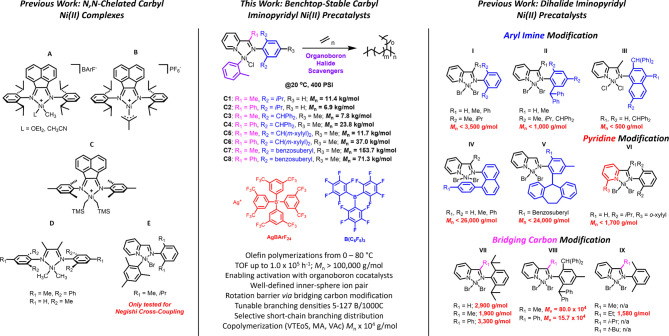
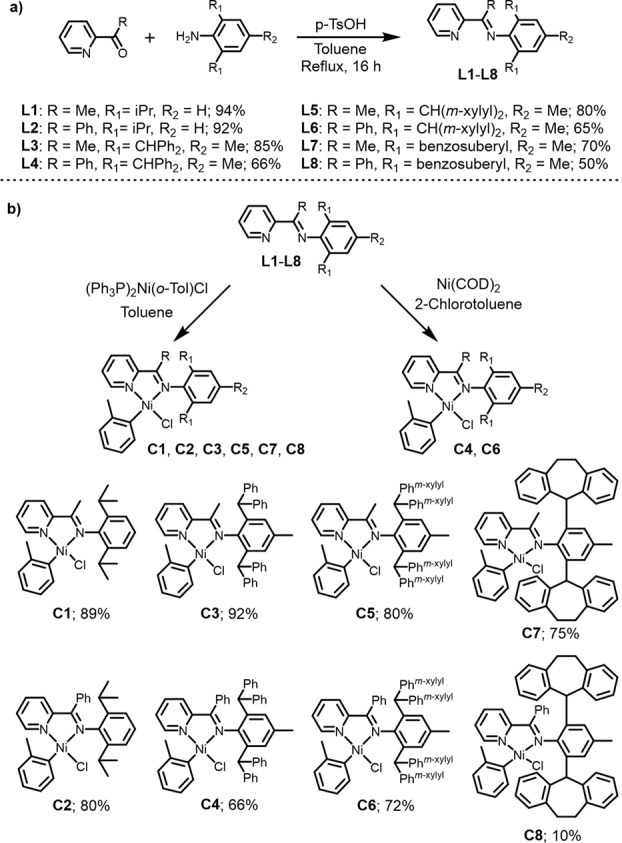
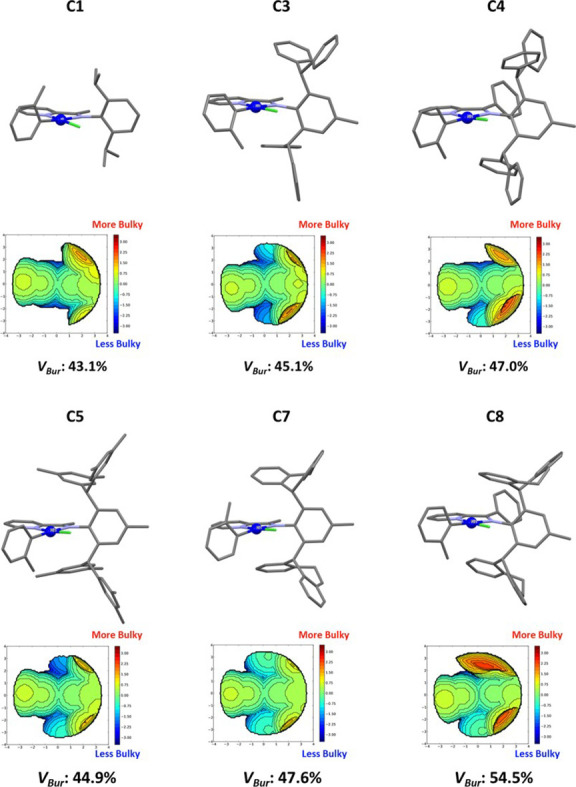
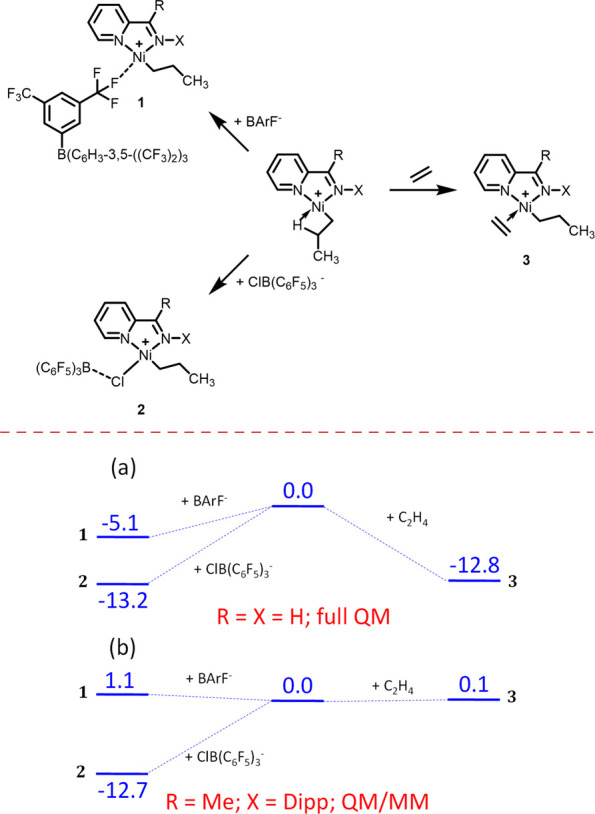
Design of catalysts for Ni-catalyzed olefin polymerization predominantly focuses on ligand design rather than the activation process when attempting to achieve a broader scope of polyolefin micro- and macrostructures. Air-stable alkyl-or aryl-functionalized Ni precatalysts were designed which eliminate the need of in situ alkylating processes and are activated solely by halide abstraction to generate the cationic complex for olefin polymerization. These complexes represent an emerging class of olefin polymerization catalysts, enabling the study of various cocatalysts forming either inner- or outer-sphere ion pairs. It is demonstrated that an organoboron cocatalyst activation produces a well-defined ion pair, which in contrast to ill-defined organoaluminum cocatalysts, can directly activate the complex by halide abstraction to yield comparatively higher molecular weight homo/copolymers. Under high ethylene pressure, broader branching densities and the gradual incorporation of short-chain branches were achieved, circumventing the need for elaborate ligand design and copolymerization with α-olefins. The underlying chain-walking mechanism and ion pair interactions were further elucidated by DFT calculations. A phenyl group on the bridging carbon functioned as a rotational barrier, producing higher molecular weight polymers compared to methyl-substituted analogs. Here, we provide a perspective to manipulate the iminopyridyl Ni system, leveraging ion pair interactions and ligand design to govern polyolefin molecular weights and microstructures.
在试图实现更广泛的聚烯烃微观和宏观结构范围时,用于镍催化烯烃聚合的催化剂设计主要集中在配体设计而非活化过程上。设计了空气稳定的烷基或芳基官能化镍预催化剂,其无需原位烷基化过程,仅通过卤化物抽取即可活化以生成用于烯烃聚合的阳离子络合物。这些络合物代表了一类新兴的烯烃聚合催化剂,能够研究形成内球或外球离子对的各种助催化剂。结果表明,有机硼助催化剂活化会产生明确的离子对,与不明确的有机铝助催化剂不同,它可通过卤化物抽取直接活化络合物,从而得到分子量相对较高的均聚物/共聚物。在高乙烯压力下,实现了更宽的支化密度和短链支化的逐步引入,无需复杂的配体设计以及与α-烯烃的共聚。通过密度泛函理论计算进一步阐明了潜在的链行走机理和离子对相互作用。桥连碳上的苯基起到旋转屏障的作用,与甲基取代的类似物相比,可生成分子量更高的聚合物。在此,我们提供了一种操控亚氨基吡啶镍体系的视角,利用离子对相互作用和配体设计来控制聚烯烃的分子量和微观结构。