Tian Yini, Ye Run, Zhang Dongmei, Zhang Yilong
Department of Tropical Diseases, Faculty of Naval Medicine, Naval Medical University, Shanghai, 200433, China.
Parasit Vectors. 2024 Dec 30;17(1):548. doi: 10.1186/s13071-024-06629-3.
The frequent communication between African and Southeast Asian (SEA) countries has led to the risk of imported malaria cases in the China-Myanmar border (CMB) region. Therefore, tracing the origins of new malaria infections is important in the maintenance of malaria-free zones in this border region. A new genotyping tool based on a robust mitochondrial (mt) /apicoplast (apico) barcode was developed to estimate genetic diversity and infer the evolutionary history of Plasmodium falciparum across the major distribution ranges. However, the mt/apico genomes of P. falciparum isolates from the CMB region to date are poorly characterized, even though this region is highly endemic to P. falciparum malaria.
We have sequenced the whole mt/apico genome of 34 CMB field isolates and utilized a published data set of 147 mt/apico genome sequences to present global genetic diversity and to revisit the evolutionary history of the CMB P. falciparum.
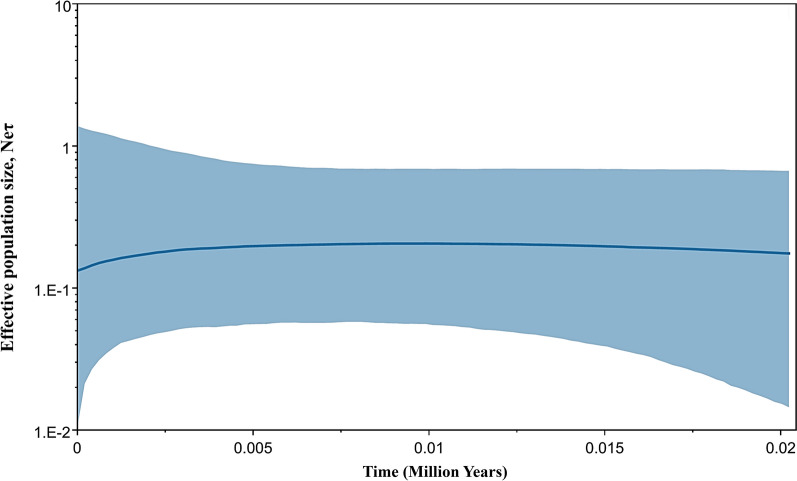
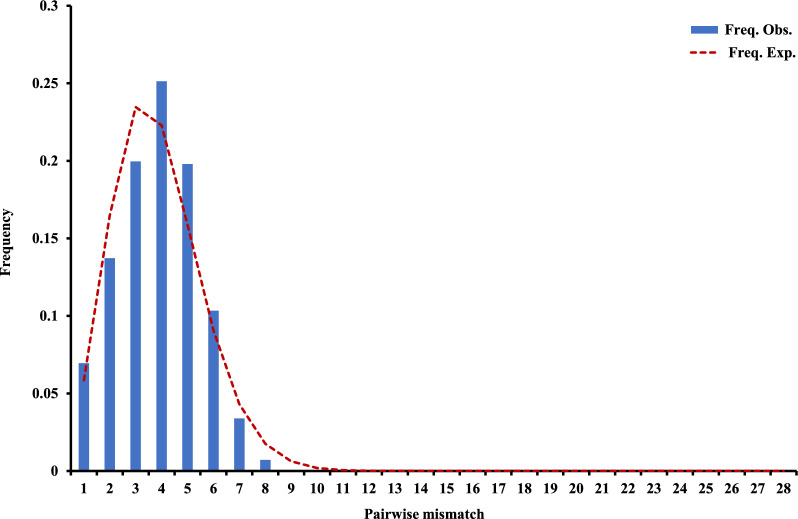
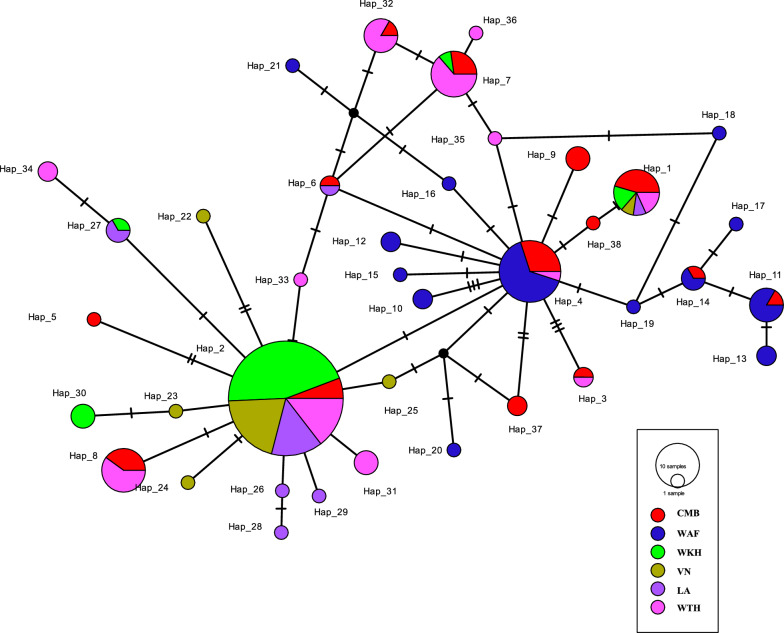
Genetic differentiation based on mt/apico genome of P. falciparum revealed that the CMB (Lazan, Myanmar) isolates presented high genetic diversity with several characteristics of ancestral populations and shared many of the genetic features with West Thailand (Mae Sot; WTH) and to some extent West African (Banjul, Gambia; Navrongo, Ghana; WAF) isolates. The reconstructed haplotype network displayed that the CMB and WTH P. falciparum isolates have the highest representation (five) in the five ancestral (central) haplotypes (H1, H2, H4, H7, and H8), which are comparatively older than isolates from other SEA populations as well as the WAF populations. In addition, the highest estimate of the time to the Most Recent Common Ancestor (TMRCA) of 42,400 (95% CI 18,300-82100) years ago was presented by the CMB P. falciparum compared to the other regional populations. The statistically significant negative values of Fu's Fs with unimodal distribution in pairwise mismatch distribution curves indicate past demographic expansions in CMB P. falciparum with slow population expansion between approximately 12,500-20,000 ybp.
The results on the complete mt/apico genome sequence analysis of the CMB P. falciparum indicated high genetic diversity with ancient population expansion and TMRCA, and it seems probable that P. falciparum might have existed in CMB, WTH, and WAF for a long time before being introduced into other Southeast Asian countries or regions. To reduce the impact of sample size or geographic bias on the estimate of the evolutionary timeline, future studies need to expand the range of sample collection and ensure the representativeness of samples across geographic distributions. Additionally, by mapping global patterns of mt/apico genome polymorphism, we will gain valuable insights into the evolutionary history of P. falciparum and optimised strategies for controlling P. falciparum malaria at international borders.
非洲与东南亚国家之间频繁的交流导致中国 - 缅甸边境地区出现输入性疟疾病例的风险。因此,追踪新疟疾病原体感染的来源对于维持该边境地区的无疟疾区域至关重要。一种基于强大的线粒体(mt)/顶质体(apico)条形码的新型基因分型工具被开发出来,用于估计恶性疟原虫在主要分布范围内的遗传多样性并推断其进化历史。然而,尽管中国 - 缅甸边境地区是恶性疟原虫疟疾的高流行区,但迄今为止,该地区恶性疟原虫分离株的mt/apico基因组特征仍不清楚。
我们对34株中国 - 缅甸边境地区的野外分离株的完整mt/apico基因组进行了测序,并利用一个已发表的包含147个mt/apico基因组序列的数据集来呈现全球遗传多样性,并重新审视中国 - 缅甸边境地区恶性疟原虫的进化历史。
基于恶性疟原虫mt/apico基因组的遗传分化表明,中国 - 缅甸边境地区(缅甸拉赞)的分离株呈现出高遗传多样性,具有一些祖先群体的特征,并且与泰国西部(湄索;WTH)以及在一定程度上与西非(冈比亚班珠尔;加纳纳夫龙戈;WAF)的分离株共享许多遗传特征。重建的单倍型网络显示,中国 - 缅甸边境地区和泰国西部的恶性疟原虫分离株在五个祖先(中心)单倍型(H1、H2、H4、H7和H8)中具有最高的代表性(五个),这些单倍型比其他东南亚群体以及西非群体的分离株相对更古老。此外,与其他区域群体相比,中国 - 缅甸边境地区恶性疟原虫的最近共同祖先时间(TMRCA)的最高估计值为42,400(95%可信区间18,300 - 82,100)年前。成对错配分布曲线中具有单峰分布的Fu's Fs的统计显著负值表明,中国 - 缅甸边境地区恶性疟原虫过去经历了种群扩张,在约12,500 - 20,000年前种群扩张缓慢。
对中国 - 缅甸边境地区恶性疟原虫完整mt/apico基因组序列分析的结果表明,其具有高遗传多样性以及古老的种群扩张和最近共同祖先时间,恶性疟原虫似乎可能在中国 - 缅甸边境地区、泰国西部和西非存在了很长时间,然后才被引入其他东南亚国家或地区。为了减少样本量或地理偏差对进化时间线估计的影响,未来的研究需要扩大样本采集范围,并确保样本在地理分布上的代表性。此外,通过绘制mt/apico基因组多态性的全球模式,我们将获得有关恶性疟原虫进化历史的宝贵见解,以及在国际边境控制恶性疟原虫疟疾的优化策略。