Kim Jaebeom, Yu Young Suk, Kim Keun Il, Baek Sung Hee
Creative Research Initiatives Center for Epigenetic Code and Diseases, School of Biological Sciences, Seoul National University, Seoul, Republic of Korea.
Department of Biological Sciences, Sookmyung Women's University, Seoul, Republic of Korea.
Autophagy. 2025 Jun;21(6):1379-1381. doi: 10.1080/15548627.2025.2465404. Epub 2025 Feb 24.
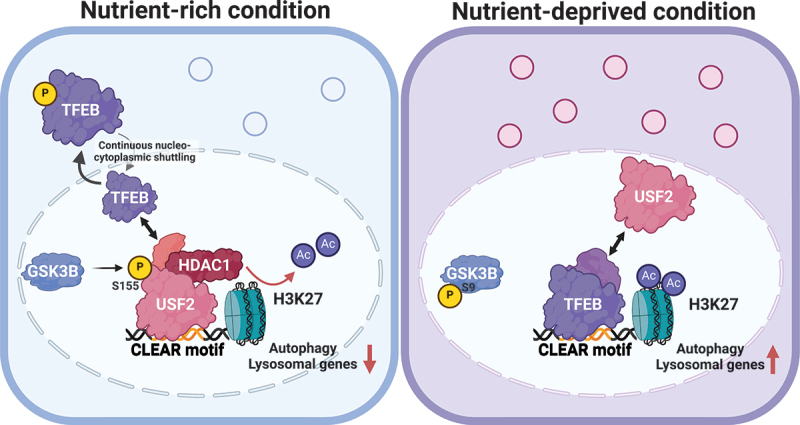
The microphthalmia/transcription factor E (MiT/TFE) family activates macroautophagy/autophagy and lysosomal genes during acute nutrient deficiency. However, the mechanisms that suppress transcription of these genes under steady-state, nutrient-rich conditions to prevent unnecessary expression remain unclear. In this study, we identified a previously unrecognized mechanism of transcriptional repression for autophagy and lysosomal genes. Under nutrient-rich conditions, USF2 (upstream transcription factor 2) binds to the coordinated lysosomal expression and regulation (CLEAR) motif, recruiting a repressive complex containing HDAC (histone deacetylase). In contrast, during nutrient deficiency, TFEB (transcription factor EB) displaces USF2 at the same motif, activating transcription. This switch is regulated by USF2 phosphorylation at serine 155 by GSK3B (glycogen synthase kinase 3 beta). Reduced phosphorylation under nutrient-deprived conditions weakens USF2's DNA binding affinity, allowing TFEB to competitively bind and activate target genes. Knockdown or knockout of upregulates specific autophagy and lysosomal genes, leading to enhanced lysosomal functionality and increased autophagic flux. In USF2-deficient cells, the SERPINA1 Z variant/antitrypsin Z - an aggregation-prone mutant protein used as a model - is rapidly cleared via the autophagy-lysosome pathway. Therefore, modulation of USF2 activity may be a therapeutic strategy for managing diseases associated with autophagy and lysosomal dysfunction.: CLEAR: coordinated lysosomal expression and regulation; GSK3B: glycogen synthase kinase 3 beta; HDAC: histone deacetylase; MiT/TFE: microphthalmia/transcription factor E; NuRD: nucleosome remodeling and deacetylation; SERPINA1 Z variant/ATZ/antitrypsin Z; TFE3: transcription factor E3; TFEB: transcription factor EB; USF2: upstream transcription factor 2.
小眼畸形/转录因子E(MiT/TFE)家族在急性营养缺乏时激活巨自噬/自噬和溶酶体基因。然而,在稳态、营养丰富的条件下抑制这些基因转录以防止不必要表达的机制仍不清楚。在本研究中,我们发现了一种以前未被认识的自噬和溶酶体基因转录抑制机制。在营养丰富的条件下,上游转录因子2(USF2)与协调溶酶体表达和调控(CLEAR)基序结合,募集一个包含组蛋白脱乙酰酶(HDAC)的抑制复合物。相反,在营养缺乏时,转录因子EB(TFEB)在同一基序上取代USF2,激活转录。这种转换由糖原合酶激酶3β(GSK3B)对USF2丝氨酸155位点的磷酸化调节。营养缺乏条件下磷酸化减少会削弱USF2的DNA结合亲和力,使TFEB能够竞争性结合并激活靶基因。敲低或敲除USF2会上调特定自噬和溶酶体基因,导致溶酶体功能增强和自噬通量增加。在缺乏USF2的细胞中,丝氨酸蛋白酶抑制剂A1 Z变体/抗胰蛋白酶Z(一种易聚集的突变蛋白,用作模型)通过自噬-溶酶体途径迅速清除。因此,调节USF2活性可能是治疗与自噬和溶酶体功能障碍相关疾病的一种策略。:CLEAR:协调溶酶体表达和调控;GSK3B:糖原合酶激酶3β;HDAC:组蛋白脱乙酰酶;MiT/TFE:小眼畸形/转录因子E;核小体重塑和脱乙酰化(NuRD);丝氨酸蛋白酶抑制剂A1 Z变体/抗胰蛋白酶Z(SERPINA1 Z variant/ATZ);转录因子E3(TFE3);转录因子EB(TFEB);上游转录因子2(USF2)