Cif Laura, Demailly Diane, Lin Jean-Pierre, Barwick Katy E, Sa Mario, Abela Lucia, Malhotra Sony, Chong Wui K, Steel Dora, Sanchis-Juan Alba, Ngoh Adeline, Trump Natalie, Meyer Esther, Vasques Xavier, Rankin Julia, Allain Meredith W, Applegate Carolyn D, Isfahani Sanaz Attaripour, Baleine Julien, Balint Bettina, Bassetti Jennifer A, Baple Emma L, Bhatia Kailash P, Blanchet Catherine, Burglen Lydie, Cambonie Gilles, Seng Emilie Chan, Bastaraud Sandra Chantot, Cyprien Fabienne, Coubes Christine, d'Hardemare Vincent, Doja Asif, Dorison Nathalie, Doummar Diane, Dy-Hollins Marisela E, Farrelly Ellyn, Fitzpatrick David R, Fearon Conor, Fieg Elizabeth L, Fogel Brent L, Forman Eva B, Fox Rachel G, Gahl William A, Galosi Serena, Gonzalez Victoria, Graves Tracey D, Gregory Allison, Hallett Mark, Hasegawa Harutomo, Hayflick Susan J, Hamosh Ada, Hully Marie, Jansen Sandra, Jeong Suh Young, Krier Joel B, Krystal Sidney, Kumar Kishore R, Laurencin Chloé, Lee Hane, Lesca Gaetan, François Laurence Lion, Lynch Timothy, Mahant Neil, Martinez-Agosto Julian A, Milesi Christophe, Mills Kelly A, Mondain Michel, Morales-Briceno Hugo, Ostergaard John R, Pal Swasti, Pallais Juan C, Pavillard Frédérique, Perrigault Pierre-Francois, Petersen Andrea K, Polo Gustavo, Poulen Gaetan, Rinne Tuula, Roujeau Thomas, Rogers Caleb, Roubertie Agathe, Sahagian Michelle, Schaefer Elise, Selim Laila, Selway Richard, Sharma Nutan, Signer Rebecca, Soldatos Ariane G, Stevenson David A, Stewart Fiona, Tchan Michel, Verma Ishwar C, de Vries Bert B A, Wilson Jenny L, Wong Derek A, Zaitoun Raghda, Zhen Dolly, Znaczko Anna, Dale Russell C, de Gusmão Claudio M, Friedman Jennifer, Fung Victor S C, King Mary D, Mohammad Shekeeb S, Rohena Luis, Waugh Jeff L, Toro Camilo, Raymond F Lucy, Topf Maya, Coubes Philippe, Gorman Kathleen M, Kurian Manju A
Département de Neurochirurgie, Unité des Pathologies Cérébrales Résistantes, Unité de Recherche sur les Comportements et Mouvements Anormaux, Hôpital Gui de Chauliac, Centre Hospitalier Régional Montpellier, Montpellier, France.
Faculté demédecine, Université de Montpellier, France.
ArXiv. 2025 Feb 10:arXiv:2502.06320v1.
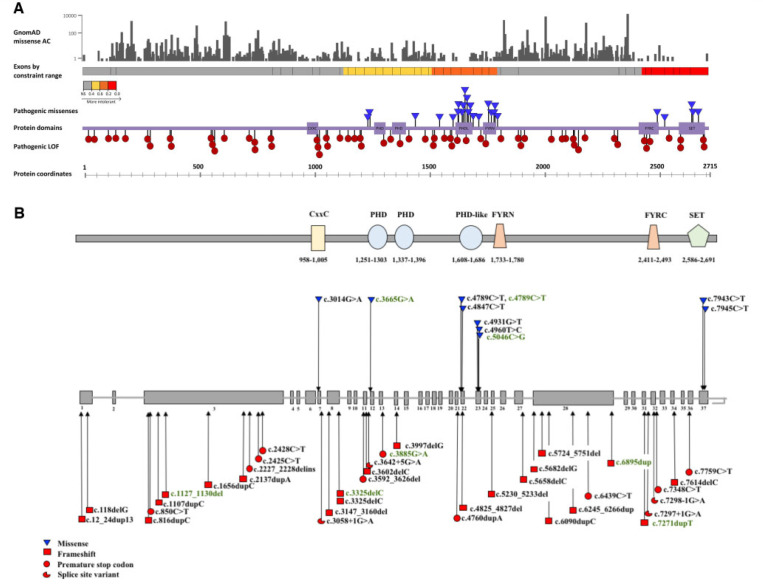
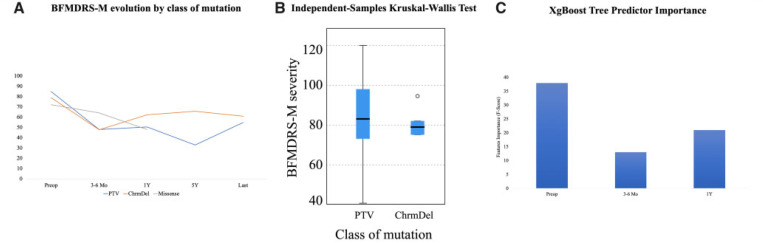
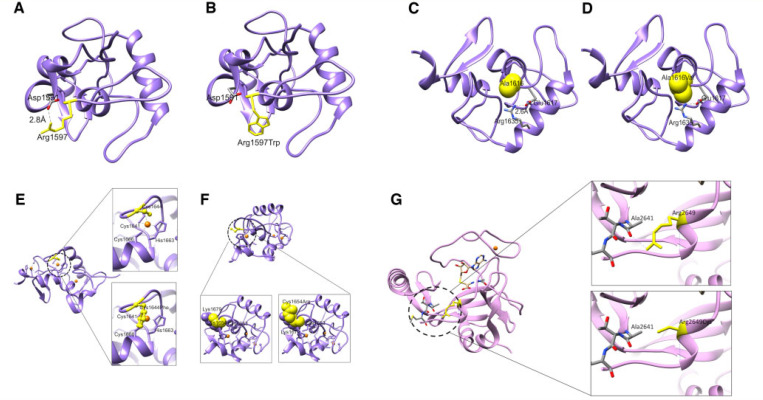
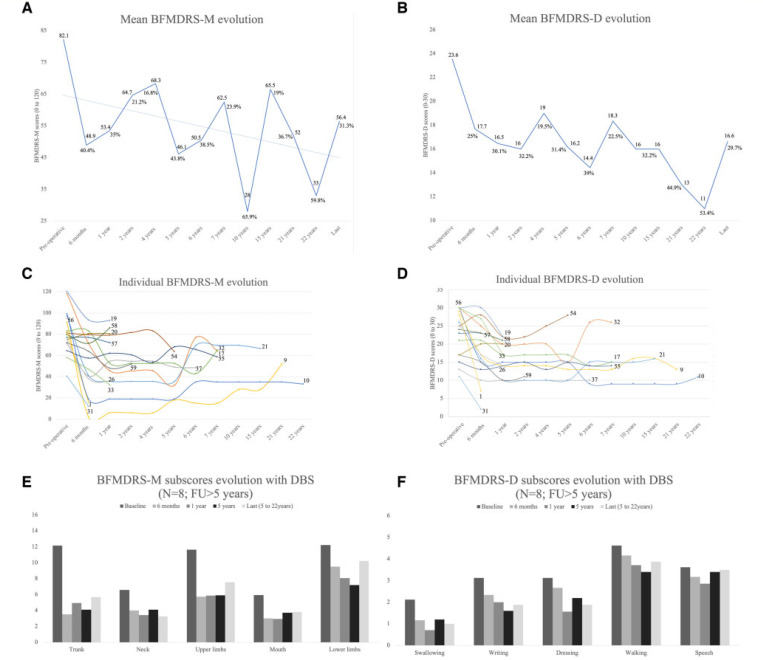
Heterozygous mutations in are associated with an early-onset, progressive and often complex dystonia (DYT28). Key characteristics of typical disease include focal motor features at disease presentation, evolving through a caudocranial pattern into generalized dystonia, with prominent oromandibular, laryngeal and cervical involvement. Although -related disease is emerging as one of the most common causes of early-onset genetic dystonia, much remains to be understood about the full spectrum of the disease. We describe a cohort of 53 patients with mutations, with detailed delineation of their clinical phenotype and molecular genetic features. We report new disease presentations, including atypical patterns of dystonia evolution and a subgroup of patients with a non-dystonic neurodevelopmental phenotype. In addition to the previously reported systemic features, our study has identified co-morbidities, including the risk of status dystonicus, intrauterine growth retardation, and endocrinopathies. Analysis of this study cohort ( = 53) in tandem with published cases ( = 80) revealed that patients with chromosomal deletions and protein truncating variants had a significantly higher burden of systemic disease (with earlier onset of dystonia) than those with missense variants. Eighteen individuals had detailed longitudinal data available after insertion of deep brain stimulation for medically refractory dystonia. Median age at deep brain stimulation was 11.5 years (range: 4.5-37.0 years). Follow-up after deep brain stimulation ranged from 0.25 to 22 years. Significant improvement of motor function and disability (as assessed by the Burke Fahn Marsden's Dystonia Rating Scales, BFMDRS-M and BFMDRS-D) was evident at ł months, 1 year and last follow-up (motor, = 0.001, = 0.004, and = 0.012; disability, = 0.009, = 0.002 and = 0.012). At 1 year post-deep brain stimulation, >50% of subjects showed BFMDRS-M and BFMDRS-D improvements of >30%. In the long-term deep brain stimulation cohort (deep brain stimulation inserted for >5 years, = 8), improvement of >30% was maintained in 5/8 and 3/8 subjects for the BFMDRS-M and BFMDRS-D, respectively. The greatest BFMDRS-M improvements were observed for trunk (53.2%) and cervical (50.5%) dystonia, with less clinical impact on laryngeal dystonia. Improvements in gait dystonia decreased from 20.9% at 1 year to 1ł.2% at last assessment; no patient maintained a fully independent gait. Reduction of BFMDRS-D was maintained for swallowing (52.9%). Five patients developed mild parkinsonism following deep brain stimulation. related disease comprises an expanding continuum from infancy to adulthood, with early evidence of genotype-phenotype correlations. Except for laryngeal dysphonia, deep brain stimulation provides a significant improvement in quality of life and function with sustained clinical benefit depending on symptoms distribution.
[基因名称]的杂合突变与早发性、进行性且通常复杂的肌张力障碍(DYT28)相关。典型疾病的关键特征包括疾病初发时的局灶性运动特征,通过尾颅模式发展为全身性肌张力障碍,伴有明显的口下颌、喉部和颈部受累。尽管[基因名称]相关疾病正逐渐成为早发性遗传性肌张力障碍最常见的病因之一,但关于该疾病的全貌仍有许多有待了解之处。我们描述了一组53例携带[基因名称]突变的患者,详细阐述了他们的临床表型和分子遗传学特征。我们报告了新的疾病表现,包括肌张力障碍演变的非典型模式以及一组具有非肌张力障碍性神经发育表型的患者。除了先前报道的全身特征外,我们的研究还发现了共病情况,包括肌张力障碍状态风险、宫内生长迟缓及内分泌疾病。将该研究队列(n = 53)与已发表病例(n = 80)进行串联分析发现,与错义变异患者相比,染色体缺失和蛋白质截短变异患者的全身性疾病负担显著更高(肌张力障碍发病更早)。18例患者在植入脑深部刺激器治疗药物难治性肌张力障碍后有详细的纵向数据。脑深部刺激时的中位年龄为11.5岁(范围:4.5 - 37.0岁)。脑深部刺激后的随访时间为0.25至22年。在1个月、1年及末次随访时,运动功能和残疾状况(通过伯克 - 法恩 - 马斯登肌张力障碍评定量表,BFMDRS - M和BFMDRS - D评估)有显著改善(运动方面,P = 0.001、P = 0.004和P = 0.012;残疾方面,P = 0.009、P = 0.002和P = 0.012)。在脑深部刺激1年后,超过50%的受试者BFMDRS - M和BFMDRS - D改善超过30%。在长期脑深部刺激队列(脑深部刺激植入超过5年,n = 8)中,BFMDRS - M和BFMDRS - D分别有5/8和3/8的受试者维持改善超过30%。观察到躯干(53.2%)和颈部(50.5%)肌张力障碍的BFMDRS - M改善最大,对喉部肌张力障碍的临床影响较小。步态肌张力障碍的改善从1年时的20.9%降至末次评估时的11.2%;没有患者维持完全独立的步态。BFMDRS - D在吞咽方面的降低得以维持(52.9%)。5例患者在脑深部刺激后出现轻度帕金森综合征。[基因名称]相关疾病涵盖了从婴儿期到成年期不断扩展的连续谱,早期就有基因型 - 表型相关性的证据。除了喉部发音障碍外,脑深部刺激根据症状分布可显著改善生活质量和功能,并带来持续的临床益处。