Lien Ju-Yin, Hii Lu Ann, Su Po-Hsuan, Chen Lin-Yu, Wen Kuo-Chang, Lai Hung-Cheng, Wang Yu-Chao
Institute of Biomedical Informatics, National Yang Ming Chiao Tung University, Taipei, Taiwan.
College of Health Technology, National Taipei University of Nursing and Health Sciences, Taipei, Taiwan.
Hum Genomics. 2025 May 17;19(1):56. doi: 10.1186/s40246-025-00763-4.
Ovarian cancer has the highest mortality rate among gynecological cancers, making early detection crucial, as the five-year survival rate drops from 92% with early-stage diagnosis compared to 31% with late-stage diagnosis. Current diagnostic methods such as histopathological examination and detection of cancer antigen 125 and human epididymis protein 4 biomarkers are either invasive or lack specificity and sensitivity. However, the Papanicolaou (Pap) test, which is widely used for cervical cancer screening, shows the potential for detecting ovarian cancer by identifying tumor DNA in cervical scrapings. Since aberrant DNA methylation patterns are linked to cancer progression, DNA methylation offers a promising avenue for early diagnosis. Therefore, this study aimed to develop a methylation-based machine-learning model to stratify patients with ovarian cancer from the cervical scraping samples collected via Pap test.
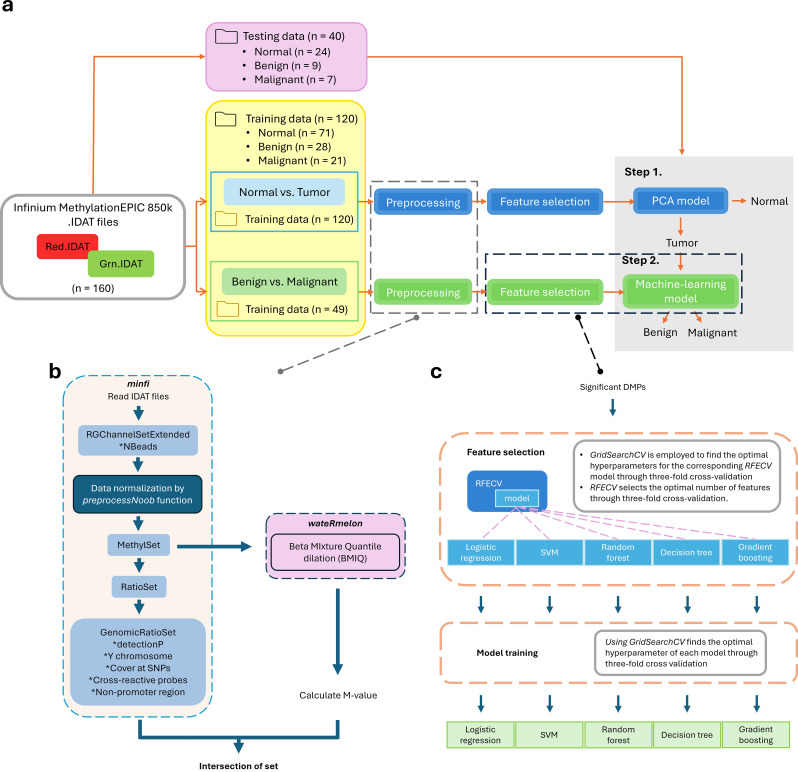
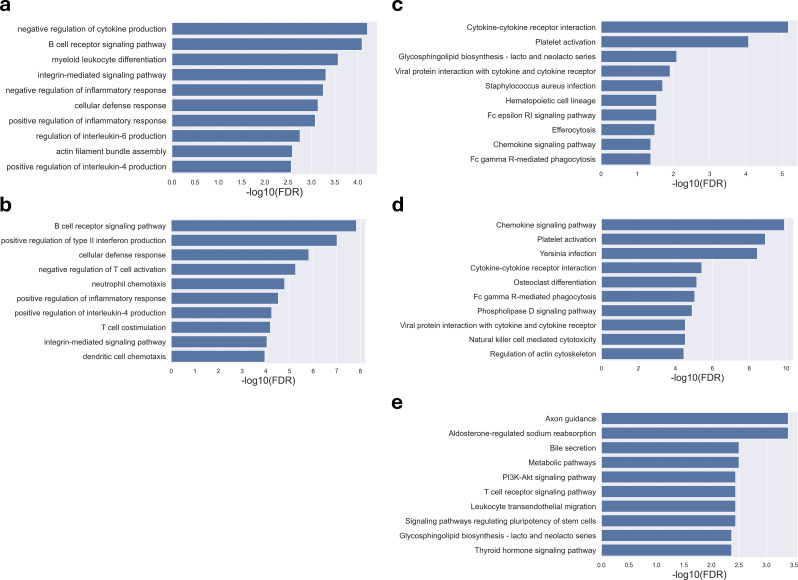
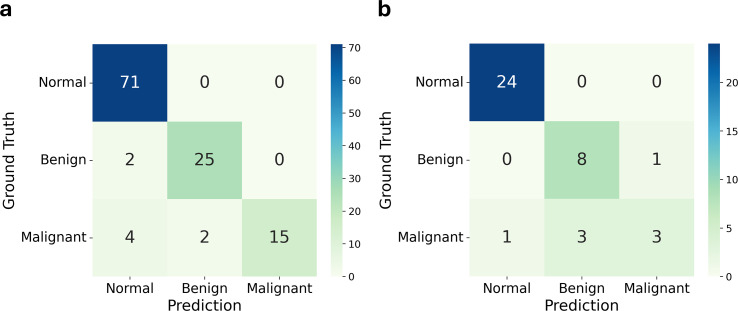
Cervical scrapings were collected by gynecologists using conventional Pap smears. In total, 160 samples were collected: 95 normal, 37 benign, and 28 malignant. Methylation data were generated using the Illumina Infinium MethylationEPIC BeadChip array, which contains approximately 850,000 CpG loci. Methylation data were initially divided into training and testing sets in a 3:1 ratio comprising 120 and 40 samples, respectively. A two-step methylation-based model was trained using the training data for classification: a principal component analysis (PCA) model, consisting of 30 features, to classify samples as normal or tumor; then a gradient boosting model, containing 16 features, to further stratify tumor samples as benign or malignant. The two-step model achieved an accuracy of 0.88 and an F1-score of 0.86 on the testing data. Furthermore, an over-representation analysis was conducted to explore the functions associated with genes mapped from differentially methylated positions (DMPs) in comparisons between normal and tumor samples, as well as between benign and malignant samples. These results suggest that DMPs may be associated with olfactory transduction when comparing normal versus tumor samples, and immune regulation when comparing benign and malignant samples.
Our two-step model shows promise for predicting ovarian cancer and suggests that cervical scrapings may be a viable alternative for sample collection during screening.
卵巢癌在妇科癌症中死亡率最高,因此早期检测至关重要,因为早期诊断的五年生存率为92%,而晚期诊断的五年生存率仅为31%。目前的诊断方法,如组织病理学检查以及检测癌抗原125和人附睾蛋白4生物标志物,要么具有侵入性,要么缺乏特异性和敏感性。然而,广泛用于宫颈癌筛查的巴氏涂片检查显示,通过识别宫颈刮片中的肿瘤DNA来检测卵巢癌具有潜力。由于异常的DNA甲基化模式与癌症进展相关,DNA甲基化提供了一条有前景的早期诊断途径。因此,本研究旨在开发一种基于甲基化的机器学习模型,以从通过巴氏涂片检查收集的宫颈刮片样本中对卵巢癌患者进行分层。
妇科医生使用传统巴氏涂片收集宫颈刮片。总共收集了160个样本:95个正常样本、37个良性样本和28个恶性样本。使用Illumina Infinium MethylationEPIC BeadChip芯片阵列生成甲基化数据,该芯片阵列包含约85万个CpG位点。甲基化数据最初以3:1的比例分为训练集和测试集,分别包含120个和40个样本。使用训练数据训练一个基于甲基化的两步模型进行分类:一个由30个特征组成的主成分分析(PCA)模型,用于将样本分类为正常或肿瘤;然后是一个包含16个特征的梯度提升模型,用于将肿瘤样本进一步分层为良性或恶性。两步模型在测试数据上的准确率为0.88,F1分数为0.86。此外,进行了过表达分析,以探索在正常样本与肿瘤样本以及良性样本与恶性样本比较中,从差异甲基化位置(DMP)映射的基因相关的功能。这些结果表明,在比较正常与肿瘤样本时,DMPs可能与嗅觉转导相关,而在比较良性与恶性样本时,DMPs可能与免疫调节相关。
我们的两步模型在预测卵巢癌方面显示出前景,并表明宫颈刮片可能是筛查期间样本采集的可行替代方法。