Zhang Hongru, Liu Lei, Shen Chuchu, Jiang Xinxue, Liu Jing, Chen Jing, Xu Senlei, Mo Yanfei
School of Acupuncture and Tuina, School of Regimen and Rehabilitation, Nanjing University of Chinese Medicine, Nanjing, China.
Dan'an College, Nanjing University of Chinese Medicine, Nanjing, China.
Research (Wash D C). 2025 May 30;8:0705. doi: 10.34133/research.0705. eCollection 2025.
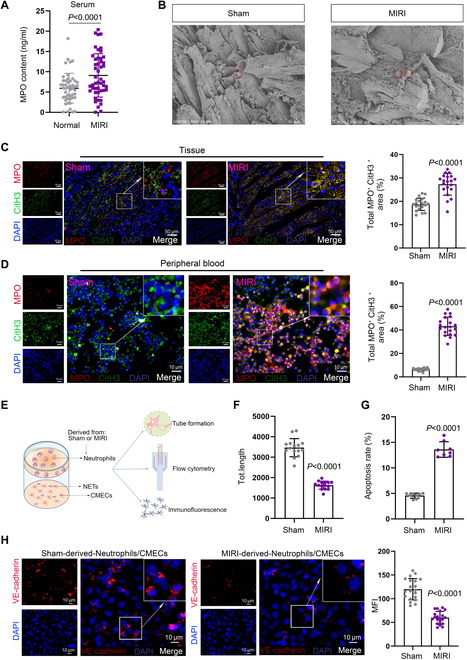
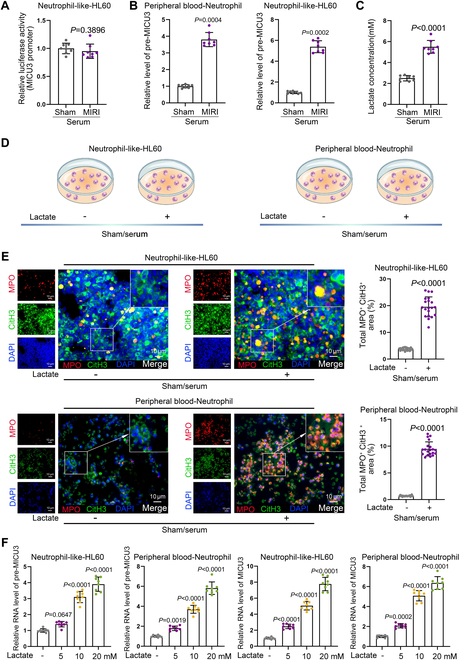
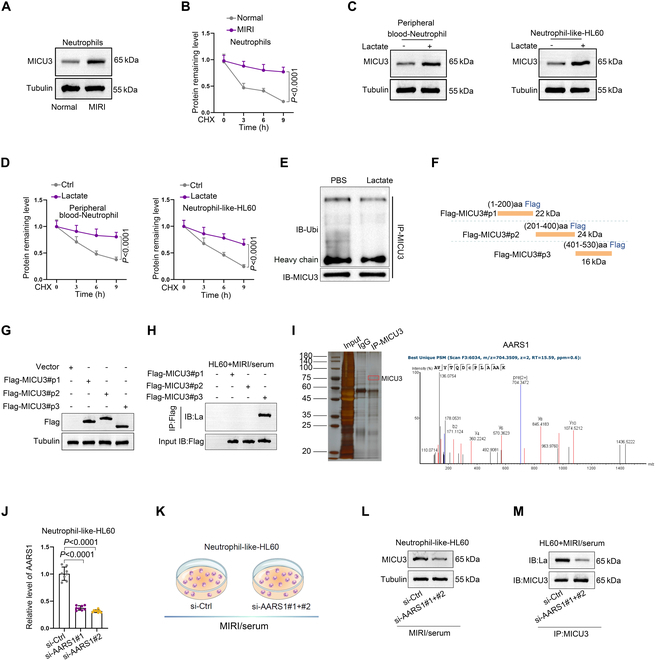
Ischemic heart disease is a leading cause of mortality and disability worldwide among cardiovascular conditions. Myocardial ischemia-reperfusion injury (MIRI) occurs following percutaneous coronary intervention, during which neutrophils generate neutrophil extracellular traps (NETs) in response to injury. This study aims to elucidate the mechanisms underlying NET activation and its impact on MIRI. Sham and MIRI rat models were established. Various techniques, including enzyme-linked immunosorbent assay, hematoxylin and eosin staining, Masson staining, and transmission electron microscopy, were used to assess endothelial cell injury and myocardial tissue inflammation. Immunofluorescence was employed to evaluate NET activation in tissues, peripheral blood neutrophils, and protein colocalization. MitoTracker and ER-Tracker staining were conducted to assess the formation of mitochondria-associated membranes (MAMs). Extracted NETs were applied to conduct microvascular endothelial cell tube formation assay and flow cytometry. RNA-sequencing and immunoprecipitation-mass spectrometry were applied to determine the key regulators. Flow cytometry and Western blot were used to assess Ca and mitophagy levels in neutrophils. Deoxyribonuclease I, NET inhibitor, was injected into MIRI rats to evaluate the in vivo effects of NET modulation on MIRI severity. MIRI was often accompanied by cardiac microvascular endothelial cell (CMEC) injury and inflammation. Lactate mediated H3K18 lactylation at the MICU3 promoter in neutrophils, enhancing its transcription and leading to elevated MICU3 levels. Besides, lactate also promoted the interaction between MICU3 and AASR1, stabilizing MICU3 through lactylation. Up-regulated MICU3 interacted with VDAC1, facilitating MAM formation, excessive Ca uptake, mitochondrial dysfunction, mitophagy activation, and NET activation. Elevated NET level exacerbated CMEC dysfunction, further aggravating MIRI. Lactate-driven MICU3 transcriptional activation and stabilization facilitates NET formation, contributing to MIRI development.
缺血性心脏病是全球心血管疾病中导致死亡和残疾的主要原因。心肌缺血再灌注损伤(MIRI)发生在经皮冠状动脉介入治疗后,在此过程中,中性粒细胞会因损伤而产生中性粒细胞胞外陷阱(NETs)。本研究旨在阐明NET激活的潜在机制及其对MIRI的影响。建立了假手术和MIRI大鼠模型。采用多种技术,包括酶联免疫吸附测定、苏木精-伊红染色、Masson染色和透射电子显微镜,来评估内皮细胞损伤和心肌组织炎症。采用免疫荧光法评估组织、外周血中性粒细胞中的NET激活以及蛋白质共定位。进行MitoTracker和ER-Tracker染色以评估线粒体相关膜(MAMs)的形成。应用提取的NETs进行微血管内皮细胞管形成试验和流式细胞术。应用RNA测序和免疫沉淀-质谱法确定关键调节因子。采用流式细胞术和蛋白质印迹法评估中性粒细胞中的钙和线粒体自噬水平。将NET抑制剂脱氧核糖核酸酶I注入MIRI大鼠体内,以评估NET调节对MIRI严重程度的体内影响。MIRI常伴有心脏微血管内皮细胞(CMEC)损伤和炎症。乳酸介导中性粒细胞中MICU3启动子处的组蛋白H3K18乳酸化,增强其转录并导致MICU3水平升高。此外,乳酸还促进MICU3与AASR1之间的相互作用,通过乳酸化使MICU3稳定。上调的MICU3与VDAC1相互作用,促进MAM形成、过量钙摄取、线粒体功能障碍、线粒体自噬激活和NET激活。NET水平升高加剧了CMEC功能障碍,进一步加重了MIRI。乳酸驱动的MICU3转录激活和稳定促进了NET形成,导致MIRI发展。