Müller Peter C, Reitz Linda S, Hemker David, Dronskowski Richard
Chair of Solid-State and Quantum Chemistry, Institute of Inorganic Chemistry, RWTH Aachen University D-52056 Aachen Germany
Chem Sci. 2025 Jun 9;16(27):12212-12226. doi: 10.1039/d5sc02936h. eCollection 2025 Jul 10.
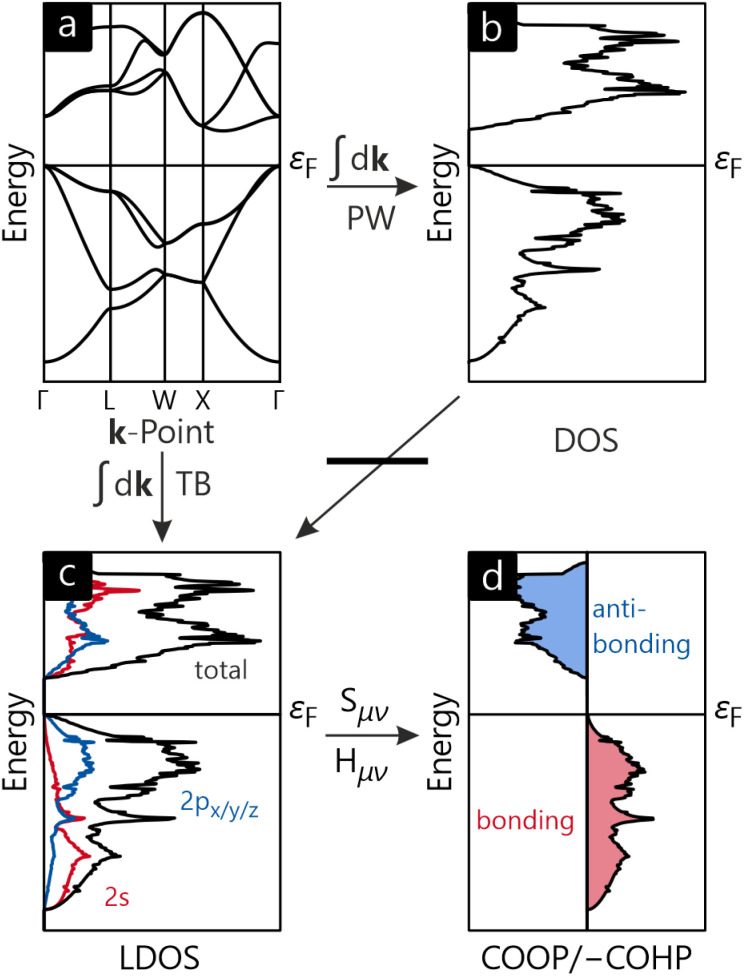
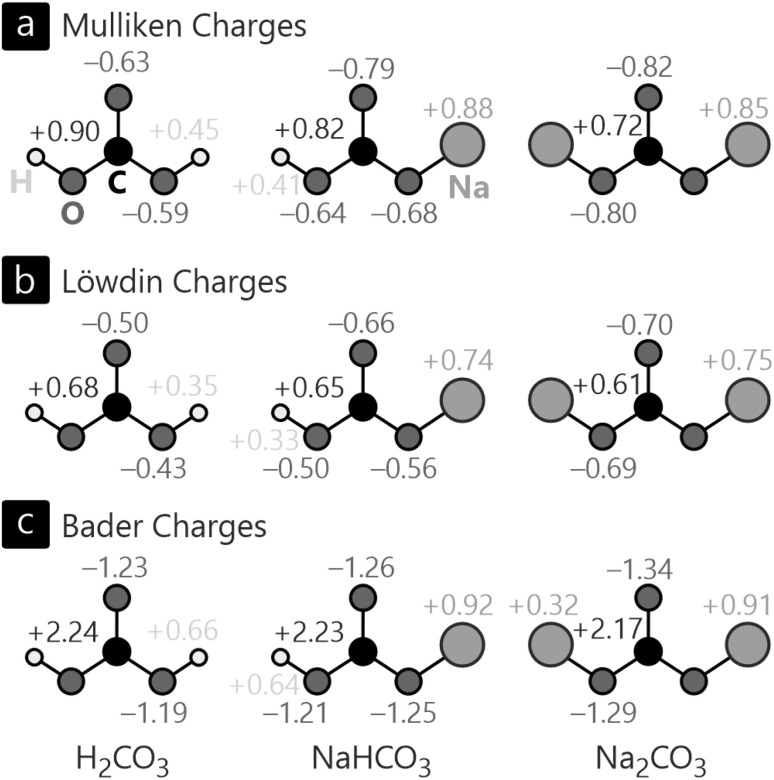
As of today, there is certainly no doubt about the quantum character of the atomistic world, most straightforwardly calculated by using wave mechanics and Schrödinger's fundamental equation from 1926. Even though one century has passed, the paramount importance of the wave function, which determines everything down to the last detail, remains unchanged, and the wave function is most conveniently approximated by a combination of orbitals, one-electron wave functions for atoms, molecules, and also solids. And it is precisely this "orbital basis" that serves as a gateway to understanding the very interactions that cause atoms to condense into solids, just like for molecules. The analysis of quantum-chemical interactions and the nature of the chemical bonding between atoms in solids by use of orbitals will be our topic in this perspective, starting with the glorious past, going over to the current practice and, of course, the magnificent prospects for the future. As electronic structures for periodic solids are most often calculated using plane waves (instead of orbitals), for simple reasons of translational symmetry and Bloch's fundamental theorem, a unitary transformation to atomic or molecular orbitals is needed for final inspection, technically solved by the LOBSTER quantum-chemistry package. LOBSTER allows for the calculation of wave function-based atomic charges, various population analyses and periodic bonding indicators, first-principles bond orders, two- and multi-centre bonding analysis, fragment-molecular analysis, and a lot more. All those techniques are illustrated from three solid-state systems deriving from carbonate chemistry.
截至今日,原子世界的量子特性毋庸置疑,最直接的计算方法是运用波动力学和1926年薛定谔的基本方程。尽管一个世纪已经过去,但决定一切直至最后细节的波函数的至关重要性依然未变,并且波函数最方便的近似是由轨道的组合给出,轨道是原子、分子乃至固体的单电子波函数。而正是这个“轨道基础”成为理解导致原子凝聚成固体的相互作用的关键入口,就如同对于分子一样。从这个角度来看,利用轨道分析固体中原子间的量子化学相互作用和化学键的本质将是我们的主题,从辉煌的过去开始,过渡到当前的实践,当然还有未来的宏伟前景。由于周期性固体的电子结构通常使用平面波(而非轨道)进行计算,出于平移对称性和布洛赫基本定理的简单原因,最终检查需要进行到原子或分子轨道的酉变换,这在技术上由LOBSTER量子化学软件包解决。LOBSTER允许计算基于波函数的原子电荷、各种布居分析和周期性键指标、第一性原理键级、双中心和多中心键分析、片段 - 分子分析等等。所有这些技术都通过源自碳酸盐化学的三个固态系统进行说明。