Uden Alexander, Dunsche Inga, Janciauskiene Sabina, Gräber Simon, Feng Longhua, Tamm Stephanie, Hedtfeld Silke, Stege Gesa, Jahn Kirsten, Kontsendorn Julia, Alfeis Nadine, Kühbandner Iris, Minso Rebecca, Dopfer Christian, Griese Matthias, Sommerburg Olaf, Ringshausen Felix C, Nährlich Lutz, Hansen Gesine, Welte Tobias, Braubach Peter, Mall Marcus A, Tümmler Burkhard, Dittrich Anna-Maria, Stanke Frauke
Department for Paediatric Pneumology, Allergology and Neonatology, Hannover Medical School, Hannover, Germany; Biomedical Research in Endstage and Obstructive Lung Disease Hannover (BREATH), German Center for Lung Research (DZL), Germany.
Department for Paediatric Pneumology, Allergology and Neonatology, Hannover Medical School, Hannover, Germany.
EBioMedicine. 2025 Jul 10;118:105848. doi: 10.1016/j.ebiom.2025.105848.
Cystic fibrosis is caused by mutations of the cystic fibrosis transmembrane conductance regulator, CFTR, an epithelial anion transport protein, responsible for, inter alia, sputum viscoelasticity in the lung. We previously identified the TNF receptor superfamily 1A TNFRSF1A (TNFR1) as a genetic modifier of CFTR function and disease severity in the CF twin and sibling study population. We aimed to replicate our findings in independent cohorts, assess the role of TNFR1 for patient survival and identify functional changes associated with TNFR1 polymorphisms.
We incorporated data from three independent long-term mono- and multicentric cohorts of people with cystic fibrosis (pwCF) to confirm the previously described association of TNFR1 with CFTR function and to extend our study to include survival data for our local cohort and a pan-European cohort of pwCF. We studied TNFR1 transcripts obtained from primary airway epithelia grown as air-liquid interface cultures to address possible mechanisms involved in up-stream and down-stream effects of TNFR1.
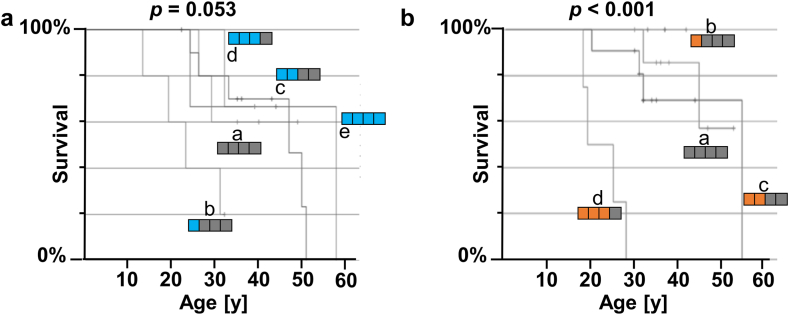
Survival differed by more than a decade when comparing carriers of contrasting TNFR1 genotypes among unrelated pwCF as well as among CF siblings pairs. The presence of the TNFR1 transcript variant TNFR1delEx2 in primary airway epithelia was associated with TNFR1 genotype.
The association of the TNFR1 transcript variant TNFR1delEx2 associates with the TNFR1 genotype, possibly mediating the genotype-survival association we found regarding TNFR1 genotype and patient survival in cystic fibrosis.
Supported by the German Ministry for Education and Research (BMBF) (82DZL009B1 to MAM and 82DZL002A1, to GH, BT, AMD, FS) and the Mukoviszidose Institut gGmbH (MI-2002, to LN, AMD, FS).
囊性纤维化由囊性纤维化跨膜传导调节因子(CFTR)突变引起,CFTR是一种上皮阴离子转运蛋白,尤其负责肺部痰液的粘弹性。在囊性纤维化双胞胎和同胞研究人群中,我们先前确定肿瘤坏死因子受体超家族1A(TNFRSF1A,TNFR1)是CFTR功能和疾病严重程度的遗传修饰因子。我们旨在在独立队列中重复我们的发现,评估TNFR1在患者生存中的作用,并确定与TNFR1多态性相关的功能变化。
我们纳入了来自三个独立的长期单中心和多中心囊性纤维化患者(pwCF)队列的数据,以确认先前描述的TNFR1与CFTR功能的关联,并将我们的研究扩展到包括我们本地队列和泛欧洲pwCF队列的生存数据。我们研究了从气液界面培养的原代气道上皮细胞中获得的TNFR1转录本,以探讨TNFR1上游和下游效应可能涉及的机制。
在不相关的pwCF以及CF同胞对中,比较具有不同TNFR1基因型的携带者时,生存期相差超过十年。原代气道上皮细胞中TNFR1转录本变体TNFR1delEx2的存在与TNFR1基因型相关。
TNFR1转录本变体TNFR1delEx2与TNFR1基因型相关,可能介导了我们在囊性纤维化中发现的TNFR1基因型与患者生存之间的基因型-生存关联。
由德国教育和研究部(BMBF)(授予MAM的82DZL009B1以及授予GH、BT、AMD、FS的82DZL002A1)和穆科维齐多斯研究所(授予LN、AMD、FS的MI-2002)资助。