Jin Ge, Fan Xiaomei, Liang Xiaoliang, Dai Honghong, Wang Jun
Department of Gynecology, the Fourth Hospital of Hebei Medical University, Shijiazhuang, China.
Department of Radiation Oncology, the Fourth Hospital of Hebei Medical University, Hebei Clinical Research Center for Radiation Oncology, Shijiazhuang, China.
Front Genet. 2025 Jul 30;16:1578075. doi: 10.3389/fgene.2025.1578075. eCollection 2025.
Abnormalities in ubiquitination-related pathways or systems are closely associated with various cancers, including cervical cancer (CC). However, the biological function and clinical value of ubiquitination-related genes (UbLGs) in CC remain unclear. This study aimed to explore key UbLGs associated with CC, construct a prognostic model, and investigate their potential clinical and immunological significance.
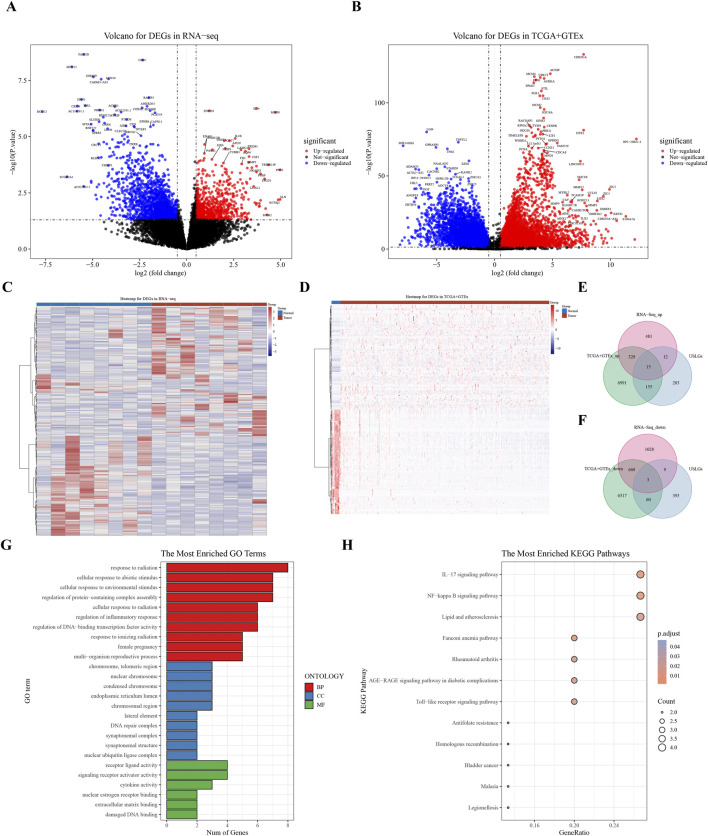
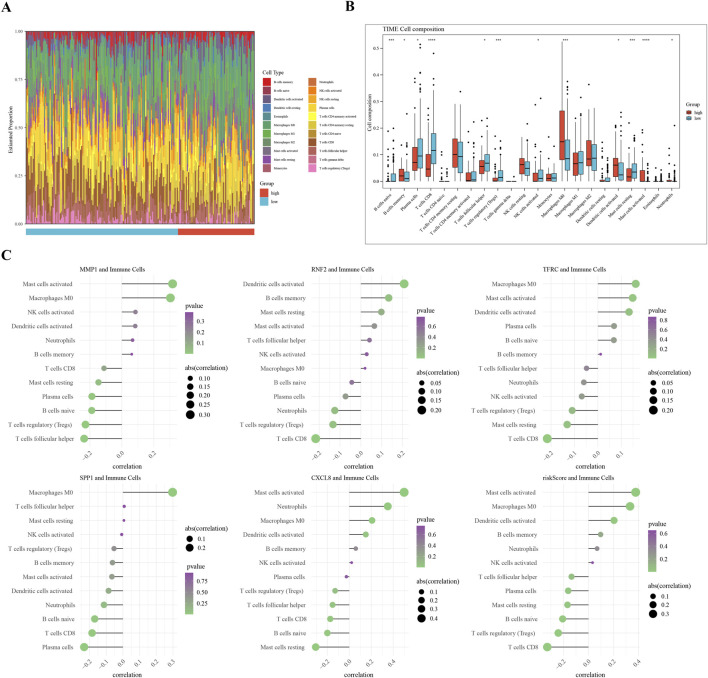
Differentially expressed genes (DEGs) between CC (tumor) and standard samples in self-sequencing and TCGA-GTEx-CESC datasets were identified using differential analysis. We identified overlaps between DEGs in both datasets and UbLGs, revealing key crossover genes. Subsequently, biological markers were identified via univariate Cox regression analysis and least absolute shrinkage and selection operator algorithms. After conducting independent prognostic analysis, immune infiltration analysis was performed to investigate the immune cells that differed between the two risk subgroups. Differences in immune checkpoint expression between the subgroups were analyzed. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR) was performed to confirm the expression trends of the biomarkers.
Differentially expressed genes related to ubiquitination were screened from the Self-seq and TCGAGTEx-CESC datasets, and five key biomarkers (MMP1, RNF2, TFRC, SPP1, and CXCL8) were identified. The risk score model constructed based on these biomarkers could effectively predict the survival rate of cervical cancer patients (AUC >0.6 for 1/3/5 years). Immune microenvironment analysis showed that 12 types of immune cells, including memory B cells and M0 macrophages, as well as four immune checkpoints, exhibited significant differences between the high-risk and low-risk groups. RT-qPCR confirmed that MMP1, TFRC, and CXCL8 were upregulated in tumor tissues.
Our study identified five ubiquitination-related biomarkers, namely, MMP1, RNF2, TFRC, SPP1, and CXCL8, which were significantly associated with CC. The validated risk model demonstrates strong predictive value for patient survival. These findings provide crucial insights into the role of ubiquitination in CC pathogenesis and offer valuable targets for advancing future research and therapeutic strategies.
泛素化相关途径或系统的异常与包括宫颈癌(CC)在内的多种癌症密切相关。然而,泛素化相关基因(UbLGs)在CC中的生物学功能和临床价值仍不清楚。本研究旨在探索与CC相关的关键UbLGs,构建预后模型,并研究其潜在的临床和免疫学意义。
使用差异分析确定自我测序以及TCGA-GTEx-CESC数据集中CC(肿瘤)与标准样本之间的差异表达基因(DEGs)。我们确定了两个数据集中DEGs与UbLGs之间的重叠,揭示了关键的交叉基因。随后,通过单变量Cox回归分析和最小绝对收缩和选择算子算法确定生物学标志物。在进行独立预后分析后,进行免疫浸润分析以研究两个风险亚组之间存在差异的免疫细胞。分析亚组之间免疫检查点表达的差异。进行实时定量聚合酶链反应(RT-qPCR)以确认生物标志物的表达趋势。
从自我测序和TCGA-GTEx-CESC数据集中筛选出与泛素化相关的差异表达基因,并鉴定出五个关键生物标志物(MMP1、RNF2、TFRC、SPP1和CXCL8)。基于这些生物标志物构建的风险评分模型可以有效预测宫颈癌患者的生存率(1/3/5年的AUC>0.6)。免疫微环境分析表明,包括记忆B细胞和M0巨噬细胞在内的12种免疫细胞以及四个免疫检查点在高风险和低风险组之间存在显著差异。RT-qPCR证实MMP1、TFRC和CXCL8在肿瘤组织中上调。
我们的研究确定了五个与泛素化相关的生物标志物,即MMP1、RNF2、TFRC、SPP1和CXCL8,它们与CC显著相关。经过验证的风险模型对患者生存具有很强的预测价值。这些发现为泛素化在CC发病机制中的作用提供了重要见解,并为推进未来的研究和治疗策略提供了有价值的靶点。