Al Z, Cohen C M
Department of Biomedical Research, St. Elizabeth's Medical Center, Boston, MA 02135.
Biochem J. 1993 Dec 15;296 ( Pt 3)(Pt 3):675-83. doi: 10.1042/bj2960675.
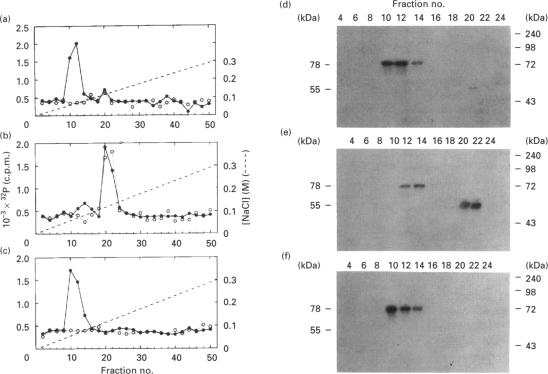
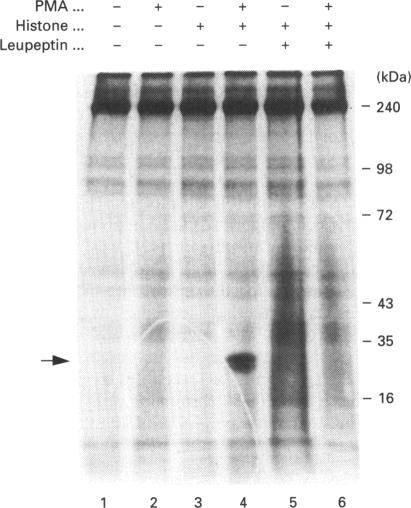
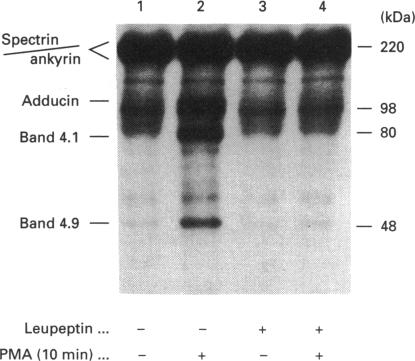
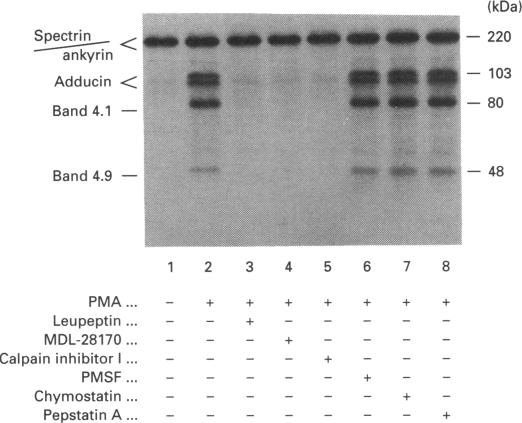
Human erythrocytes contain cytosolic protein kinase C (PKC) which, when activated by phorbol 12-myristate 13-acetate (PMA), induces the phosphorylation of the membrane skeletal proteins band 4.1, band 4.9 and adducin. We found that brief treatments of erythrocytes with PMA resulted in a decrease in cytosolic PKC content and in the transient appearance in the cytosol of a Ca(2+)- and phospholipid-independent 55 kDa fragment of PKC, called PKM. Prolonged treatment with PMA resulted in the complete and irreversible loss of erythrocyte PKC. To investigate the possible role of calpain in this process, the calpain inhibitors leupeptin and E-64 were sealed inside erythrocytes by reversible haemolysis. Both inhibitors prolonged the lifetime of PKC in PMA-treated cells, and leupeptin was shown to block the PMA-stimulated appearance of PKM in the cytosol. Significantly, leupeptin also completely blocked PMA-stimulated phosphorylation of membrane and cytosolic substrates. This effect was mimicked by other calpain inhibitors (MDL-28170 and calpain inhibitor I), but did not occur when other protease inhibitors such as phenylmethanesulphonyl fluoride, pepstatin A or chymostatin were used. In addition, the phosphorylation of exogenous histone sealed inside erythrocytes was also blocked by leupeptin. Immunoblotting showed that leupeptin did not prevent the PMA-induced translocation of PKC to the erythrocyte membrane. Thus inhibition of PKC phosphorylation of membrane skeletal proteins by calpain inhibitors was not due to inhibition of PKC translocation to the membrane. Our results suggest that PMA treatment of erythrocytes results in the translocation of PKC to the plasma membrane, followed by calpain-mediated cleavage of PKC to PKM. This cleavage, or some other leupeptin-inhibitable process, is a necessary step for the phosphorylation of membrane skeletal substrates, suggesting that the short-lived PKM may be responsible for membrane skeletal phosphorylation. Our results suggest a potential mechanism whereby erythrocyte PKC may be subject to continual down-regulation during the lifespan of the erythrocyte due to repeated activation events, possibly related to transient Ca2+ influx. Such down-regulation may play an important role in erythrocyte survival or pathophysiology.
人类红细胞含有胞质蛋白激酶C(PKC),当被佛波醇12 - 肉豆蔻酸酯13 - 乙酸酯(PMA)激活时,会诱导膜骨架蛋白4.1带、4.9带和内收蛋白的磷酸化。我们发现,用PMA对红细胞进行短暂处理会导致胞质PKC含量降低,并在胞质中短暂出现一种不依赖钙和磷脂的55 kDa的PKC片段,称为PKM。用PMA进行长时间处理会导致红细胞PKC完全且不可逆地丧失。为了研究钙蛋白酶在这一过程中可能的作用,通过可逆性溶血将钙蛋白酶抑制剂亮抑酶肽和E - 64封入红细胞内。两种抑制剂都延长了PMA处理细胞中PKC的寿命,并且亮抑酶肽被证明能阻断PMA刺激的PKM在胞质中的出现。值得注意的是,亮抑酶肽还完全阻断了PMA刺激的膜和胞质底物的磷酸化。其他钙蛋白酶抑制剂(MDL - 28170和钙蛋白酶抑制剂I)也有类似效果,但使用其他蛋白酶抑制剂如苯甲基磺酰氟、胃蛋白酶抑制剂A或抑肽酶时则不会出现这种情况。此外,封入红细胞内的外源组蛋白的磷酸化也被亮抑酶肽阻断。免疫印迹显示亮抑酶肽不会阻止PMA诱导的PKC向红细胞膜的转位。因此,钙蛋白酶抑制剂对膜骨架蛋白PKC磷酸化的抑制并非由于抑制PKC向膜的转位。我们的结果表明,用PMA处理红细胞会导致PKC转位到质膜,随后钙蛋白酶介导PKC裂解为PKM。这种裂解,或其他亮抑酶肽可抑制的过程,是膜骨架底物磷酸化的必要步骤,这表明短暂存在的PKM可能负责膜骨架的磷酸化。我们的结果提示了一种潜在机制,即由于反复的激活事件,红细胞PKC在红细胞寿命期间可能会持续下调,这可能与短暂的钙离子内流有关。这种下调可能在红细胞存活或病理生理学中起重要作用。