Hildt E, Oess S
Klinikum der Universität Ulm, Ulm, 89081 Germany.
J Exp Med. 1999 Jun 7;189(11):1707-14. doi: 10.1084/jem.189.11.1707.
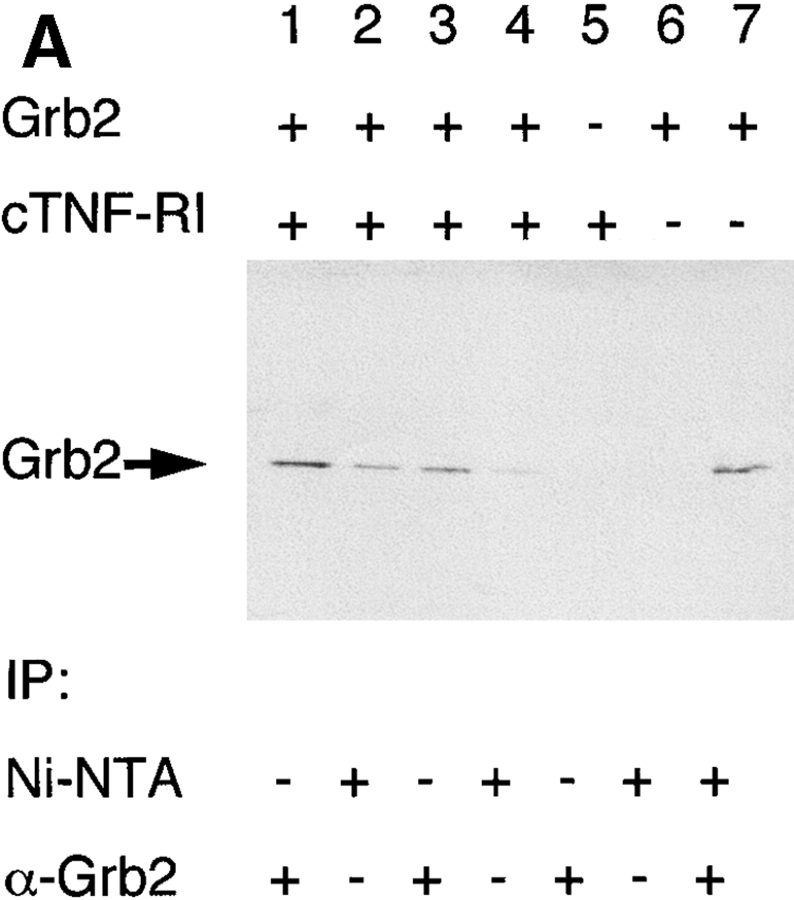
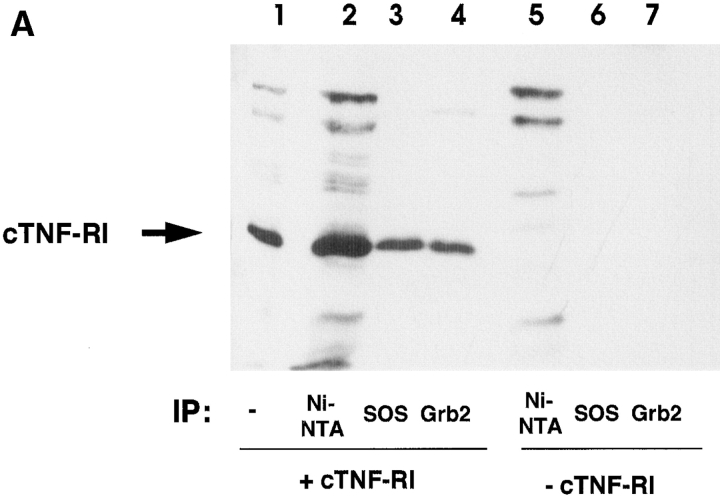
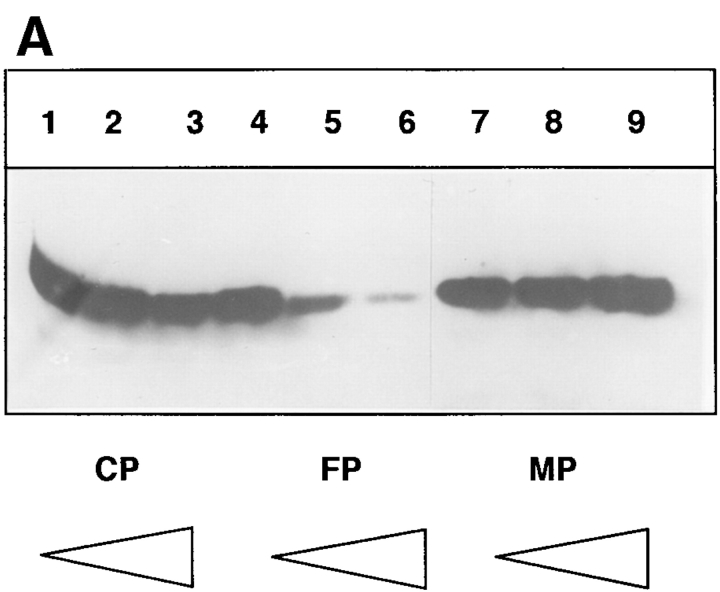
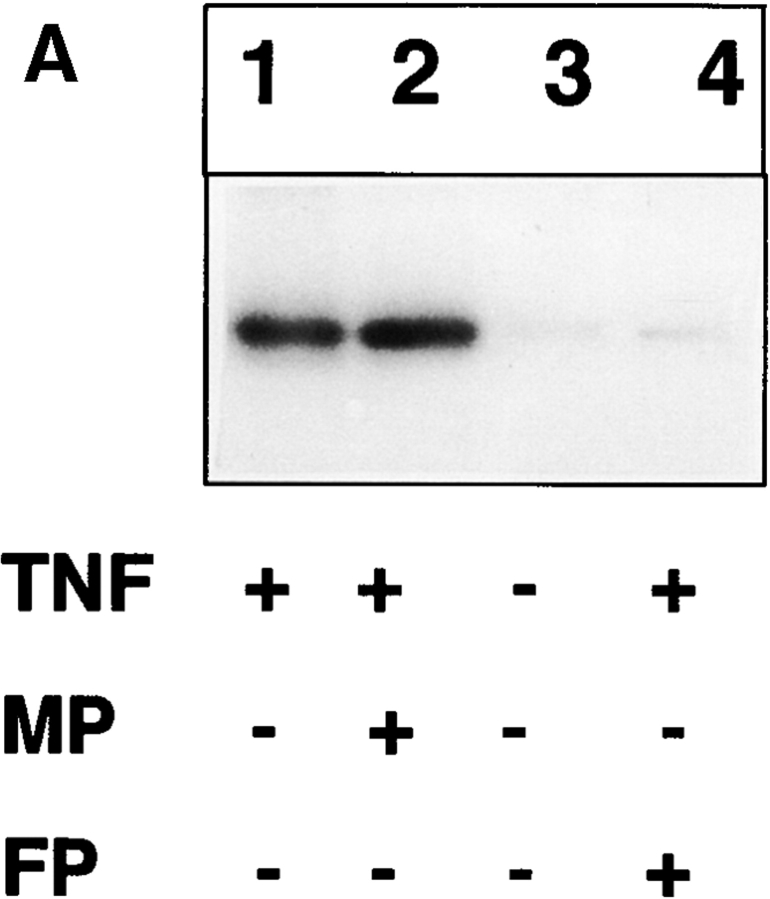
Tumor necrosis factor alpha (TNF-alpha) is a proinflammatory cytokine. Its pleiotropic biological properties are signaled through two distinct cell surface receptors: the TNF receptor type I (TNFR-I) and the TNF receptor type II. Neither of the two receptors possesses tyrosine kinase activity. A large majority of TNF-alpha-dependent activities can be mediated by TNFR-I. Recently, c-Raf-1 kinase was identified as an intracellular target of a signal transduction cascade initiated by binding of TNF-alpha to TNFR-I. However, the mechanism engaged in TNF-alpha-dependent activation of c-Raf-1 kinase is still enigmatic. Here we report that the cytosolic adapter protein Grb2 is a novel binding partner of TNFR-I. Grb2 binds with its COOH-terminal SH3 domain to a PLAP motif within TNFR-I and with its NH2-terminal SH3 domain to SOS (son of sevenless). A PLAP deletion mutant of TNFR-I fails to bind Grb2. The TNFR-I/Grb2 interaction is essential for the TNF-alpha-dependent activation of c-Raf-1 kinase; activation of c-Raf-1 kinase by TNF-alpha can be blocked by coexpression of Grb2 mutants harboring inactivating point mutations in the NH2- or COOH-terminal SH3 domain, cell-permeable peptides that disrupt the Grb2/TNFR-I interaction or transdominant negative Ras. Functionality of the TNFR-I/Grb2/SOS/Ras interaction is a prerequisite but not sufficient for TNF-alpha-dependent activation of c-Raf-1 kinase. Inhibition of the TNFR-I/FAN (factor associated with neutral sphingomyelinase) interaction, which is essential for TNF-alpha-dependent activation of the neutral sphingomyelinase, either by cell-permeable peptides or by deletion of the FAN binding domain, prevents activation of c-Raf-1 kinase. In conclusion, binding of the Grb2 adapter protein via its COOH-terminal SH3 domain to the nontyrosine kinase receptor TNFR-I results in activation of a signaling cascade known so far to be initiated, in the case of the tyrosine kinase receptors, by binding of the SH2 domain of Grb2 to phosphotyrosine.
肿瘤坏死因子α(TNF-α)是一种促炎细胞因子。其多效性生物学特性通过两种不同的细胞表面受体传递信号:I型TNF受体(TNFR-I)和II型TNF受体。这两种受体均不具有酪氨酸激酶活性。绝大多数TNF-α依赖性活性可由TNFR-I介导。最近,c-Raf-1激酶被确定为TNF-α与TNFR-I结合引发的信号转导级联反应的细胞内靶点。然而,TNF-α依赖性激活c-Raf-1激酶的机制仍然不明。在此我们报告,胞质衔接蛋白Grb2是TNFR-I的新型结合伴侣。Grb2通过其COOH末端的SH3结构域与TNFR-I内的PLAP基序结合,并通过其NH2末端的SH3结构域与SOS(七号染色体失活蛋白)结合。TNFR-I的PLAP缺失突变体无法结合Grb2。TNFR-I与Grb2的相互作用对于TNF-α依赖性激活c-Raf-1激酶至关重要;TNF-α对c-Raf-1激酶的激活可被共表达在NH2或COOH末端SH3结构域带有失活点突变的Grb2突变体、破坏Grb2/TNFR-I相互作用的细胞可渗透肽或显性负性Ras所阻断。TNFR-I/Grb2/SOS/Ras相互作用的功能性是TNF-α依赖性激活c-Raf-1激酶的前提条件,但并不充分。通过细胞可渗透肽或缺失FAN结合结构域抑制TNFR-I与FAN(与中性鞘磷脂酶相关的因子)的相互作用,而这种相互作用对于TNF-α依赖性激活中性鞘磷脂酶至关重要,可阻止c-Raf-1激酶的激活。总之,Grb2衔接蛋白通过其COOH末端的SH3结构域与非酪氨酸激酶受体TNFR-I结合,导致激活一种信号级联反应,就酪氨酸激酶受体而言,迄今为止已知该信号级联反应是由Grb2的SH2结构域与磷酸酪氨酸结合引发的。