Cifuentes-Diaz C, Frugier T, Tiziano F D, Lacène E, Roblot N, Joshi V, Moreau M H, Melki J
Molecular Neurogenetics Laboratory, Institut National de la Santé et de la Recherche Médicale (INSERM), Université d'Evry, EMI-9913, Genopole, 91057 Evry, France.
J Cell Biol. 2001 Mar 5;152(5):1107-14. doi: 10.1083/jcb.152.5.1107.
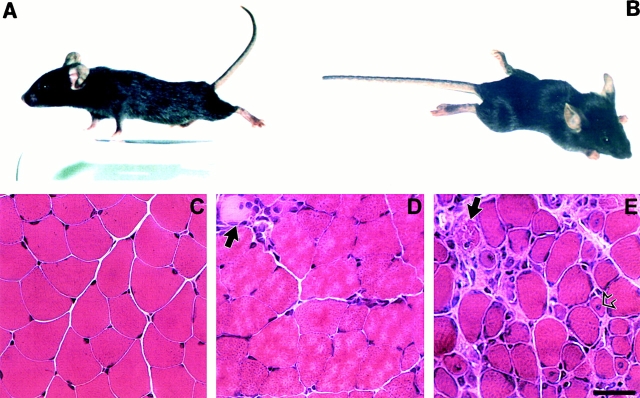
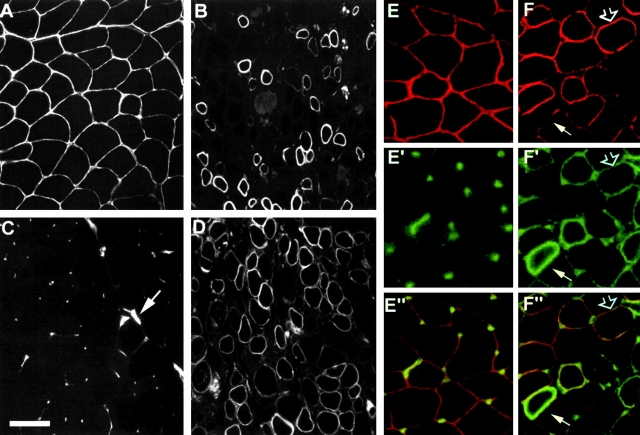
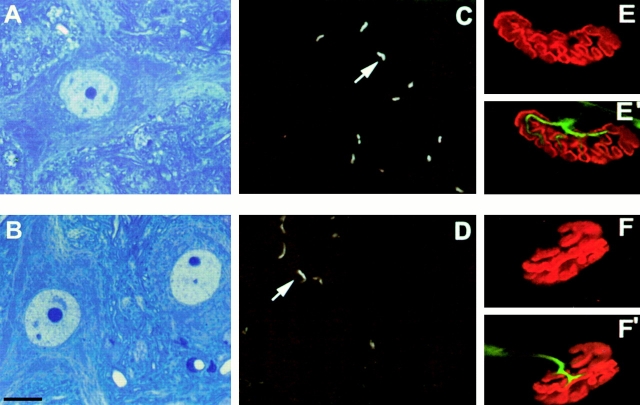
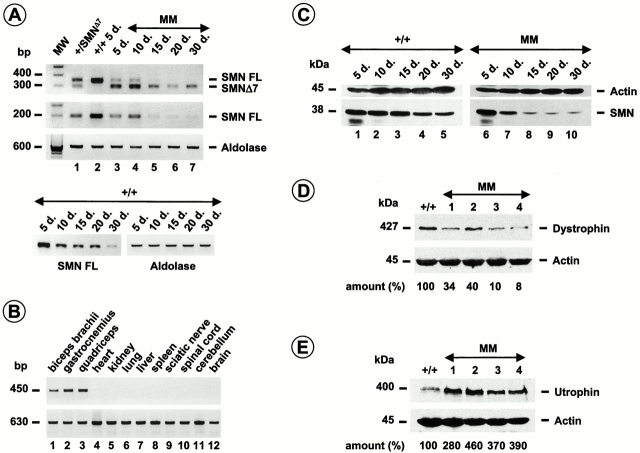
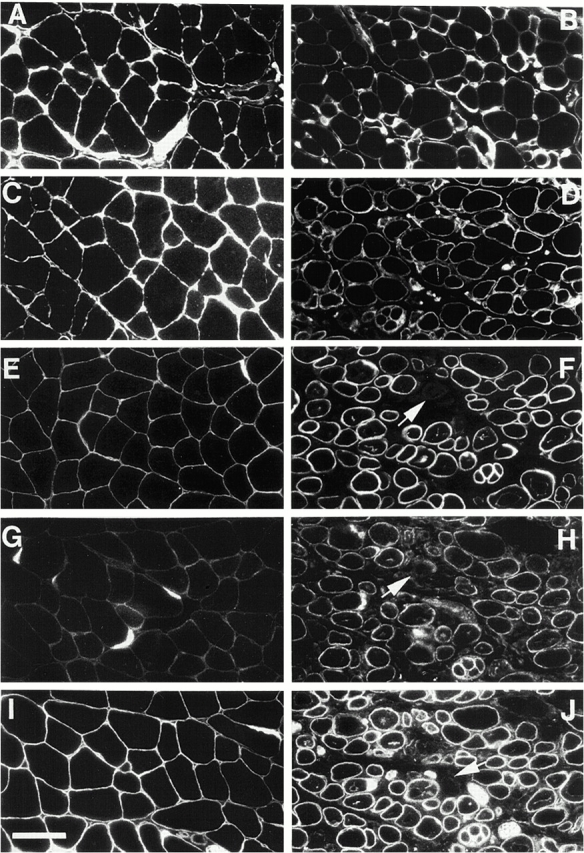
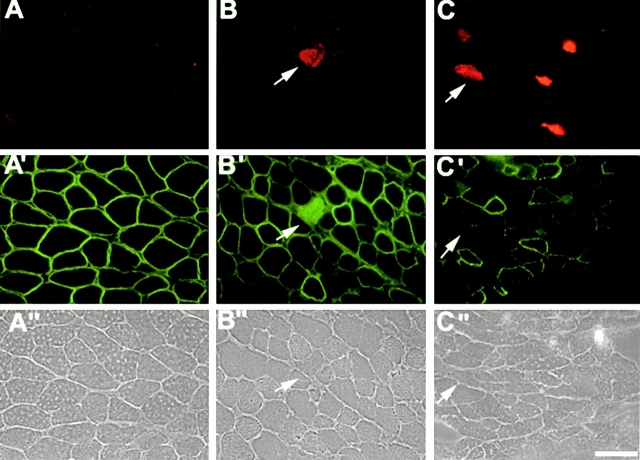
Spinal muscular atrophy (SMA) is characterized by degeneration of motor neurons of the spinal cord associated with muscle paralysis and caused by mutations of the survival motor neuron gene (SMN). To determine whether SMN gene defect in skeletal muscle might have a role in SMA pathogenesis, deletion of murine SMN exon 7, the most frequent mutation found in SMA, has been restricted to skeletal muscle by using the Cre-loxP system. Mutant mice display ongoing muscle necrosis with a dystrophic phenotype leading to muscle paralysis and death. The dystrophic phenotype is associated with elevated levels of creatine kinase activity, Evans blue dye uptake into muscle fibers, reduced amount of dystrophin and upregulation of utrophin expression suggesting a destabilization of the sarcolemma components. The mutant mice will be a valuable model for elucidating the underlying mechanism. Moreover, our results suggest a primary involvement of skeletal muscle in human SMA, which may contribute to motor defect in addition to muscle denervation caused by the motor neuron degeneration. These data may have important implications for the development of therapeutic strategies in SMA.
脊髓性肌萎缩症(SMA)的特征是脊髓运动神经元退化,伴有肌肉麻痹,由生存运动神经元基因(SMN)突变引起。为了确定骨骼肌中的SMN基因缺陷是否可能在SMA发病机制中起作用,通过使用Cre-loxP系统,将小鼠SMN外显子7(SMA中最常见的突变)的缺失限制在骨骼肌中。突变小鼠表现出持续的肌肉坏死,伴有营养不良表型,导致肌肉麻痹和死亡。营养不良表型与肌酸激酶活性水平升高、伊文思蓝染料摄取到肌纤维中、肌营养不良蛋白量减少以及抗肌萎缩蛋白表达上调有关,提示肌膜成分不稳定。突变小鼠将成为阐明潜在机制的有价值模型。此外,我们的结果表明骨骼肌在人类SMA中起主要作用,这除了运动神经元退化导致的肌肉去神经支配外,可能还导致运动缺陷。这些数据可能对SMA治疗策略的开发具有重要意义。