Muro Shizuko, Takemasa Ichiro, Oba Shigeyuki, Matoba Ryo, Ueno Noriko, Maruyama Chiyuri, Yamashita Riu, Sekimoto Mitsugu, Yamamoto Hirofumi, Nakamori Shoji, Monden Morito, Ishii Shin, Kato Kikuya
Taisho Laboratory of Functional Genomics, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara 630-0101, Japan.
Genome Biol. 2003;4(3):R21. doi: 10.1186/gb-2003-4-3-r21. Epub 2003 Feb 27.
Individual human carcinomas have distinct biological and clinical properties: gene-expression profiling is expected to unveil the underlying molecular features. Particular interest has been focused on potential diagnostic and therapeutic applications. Solid tumors, such as colorectal carcinoma, present additional obstacles for experimental and data analysis.
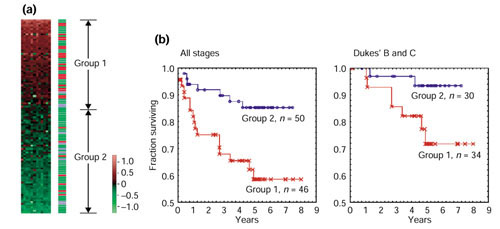
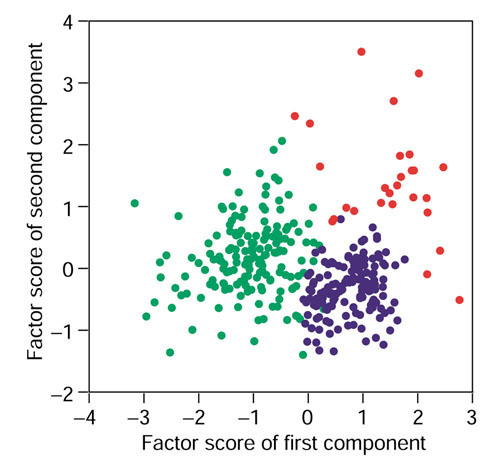
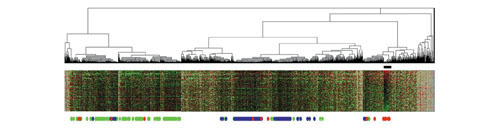
We analyzed the expression levels of 1,536 genes in 100 colorectal cancer and 11 normal tissues using adaptor-tagged competitive PCR, a high-throughput reverse transcription-PCR technique. A parametric clustering method using the Gaussian mixture model and the Bayes inference revealed three groups of expressed genes. Two contained large numbers of genes. One of these groups correlated well with both the differences between tumor and normal tissues and the presence or absence of distant metastasis, whereas the other correlated only with the tumor/normal difference. The third group comprised a small number of genes. Approximately half showed an identical expression pattern, and cancer tissues were classified into two groups by their expression levels. The high-expression group had strong correlation with distant metastasis, and a poorer survival rate than the low-expression group, indicating possible clinical applications of these genes. In addition to c-yes, a homolog of a viral oncogene, prognostic indicators included genes specific to glial cells, which gives a new link between malignancy and ectopic gene expression.
The malignancy of human colorectal carcinoma is correlated with a unique expression pattern of a specific group of genes, allowing the classification of tumor tissues into two clinically distinct groups.
个体人类癌具有独特的生物学和临床特性:基因表达谱有望揭示潜在的分子特征。人们尤其关注其潜在的诊断和治疗应用。实体瘤,如结直肠癌,在实验和数据分析方面存在额外的障碍。
我们使用衔接子标签竞争性PCR(一种高通量逆转录PCR技术)分析了100例结直肠癌组织和11例正常组织中1536个基因的表达水平。一种使用高斯混合模型和贝叶斯推理的参数聚类方法揭示了三组表达基因。其中两组包含大量基因。其中一组与肿瘤组织和正常组织之间的差异以及远处转移的有无都有很好的相关性,而另一组仅与肿瘤/正常差异相关。第三组由少数基因组成。大约一半呈现相同的表达模式,癌组织根据其表达水平被分为两组。高表达组与远处转移密切相关,生存率低于低表达组,表明这些基因可能具有临床应用价值。除了病毒癌基因的同源物c-yes外,预后指标还包括神经胶质细胞特有的基因,这在恶性肿瘤和异位基因表达之间建立了新的联系。
人类结直肠癌的恶性程度与特定一组基因的独特表达模式相关,这使得肿瘤组织能够被分为两个临床特征不同的组。