Zheng Xinglong, Nishio Kenji, Majerus Elaine M, Sadler J Evan
Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, Missouri 63110, USA.
J Biol Chem. 2003 Aug 8;278(32):30136-41. doi: 10.1074/jbc.M305331200. Epub 2003 Jun 5.
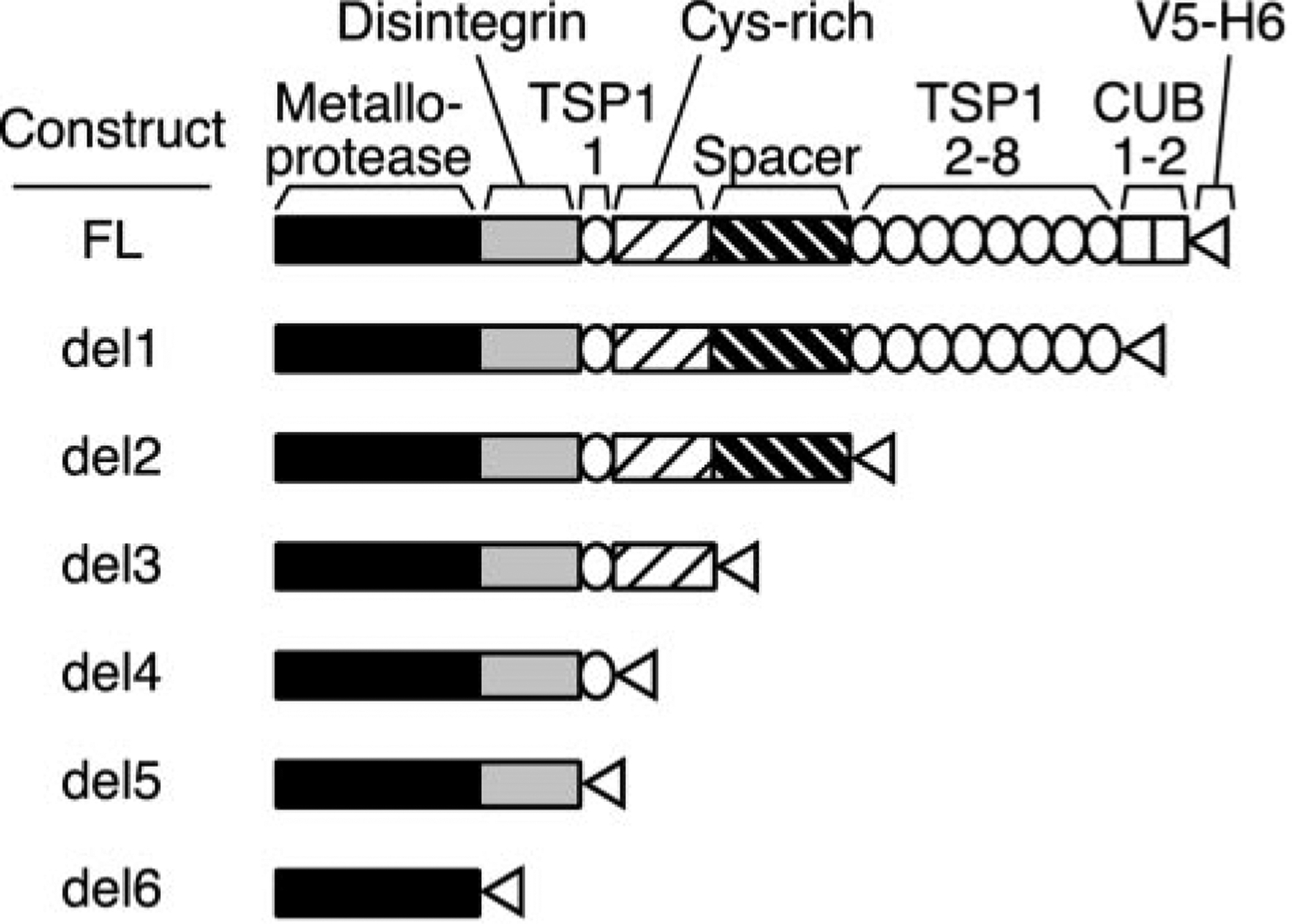
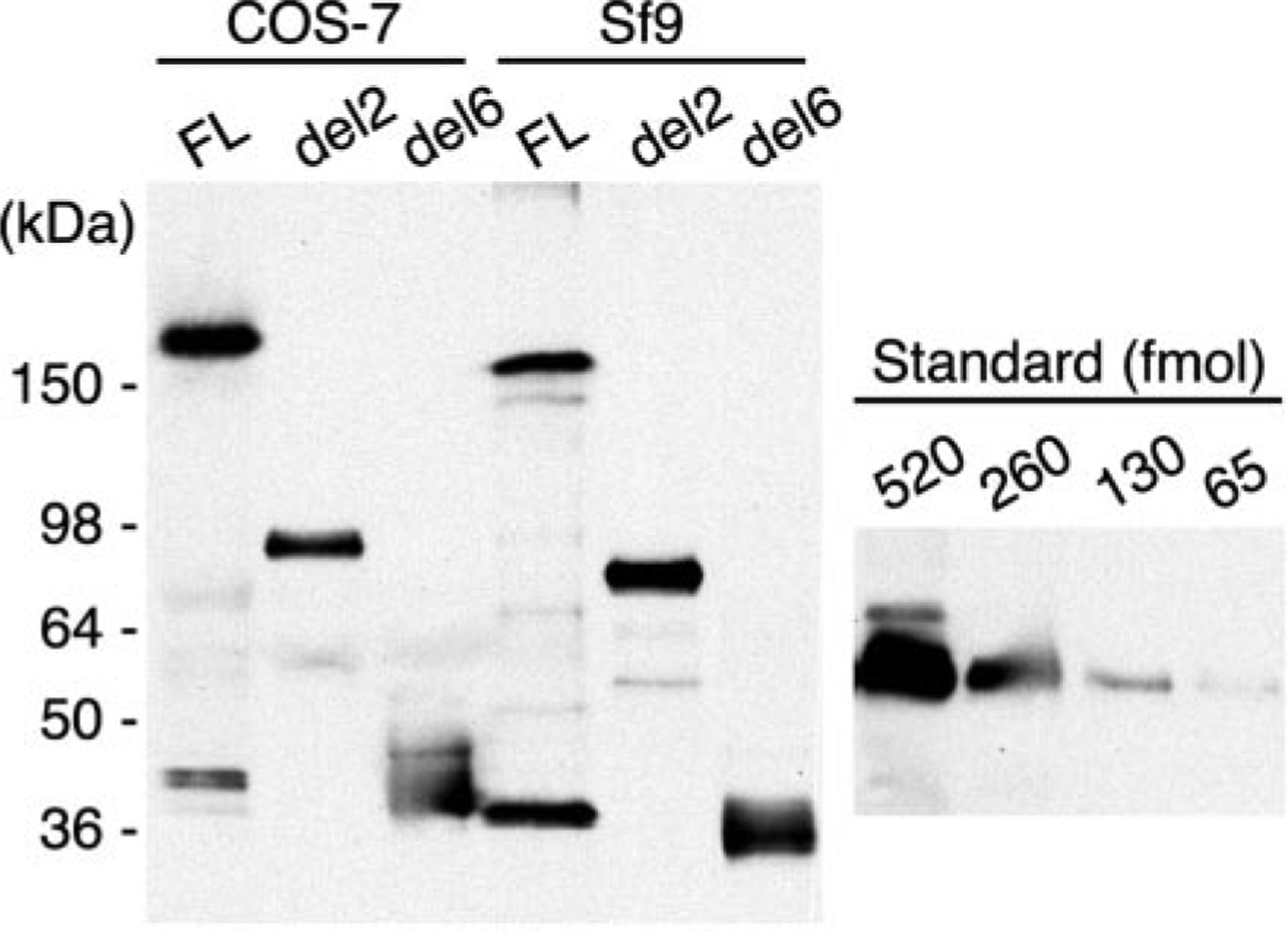
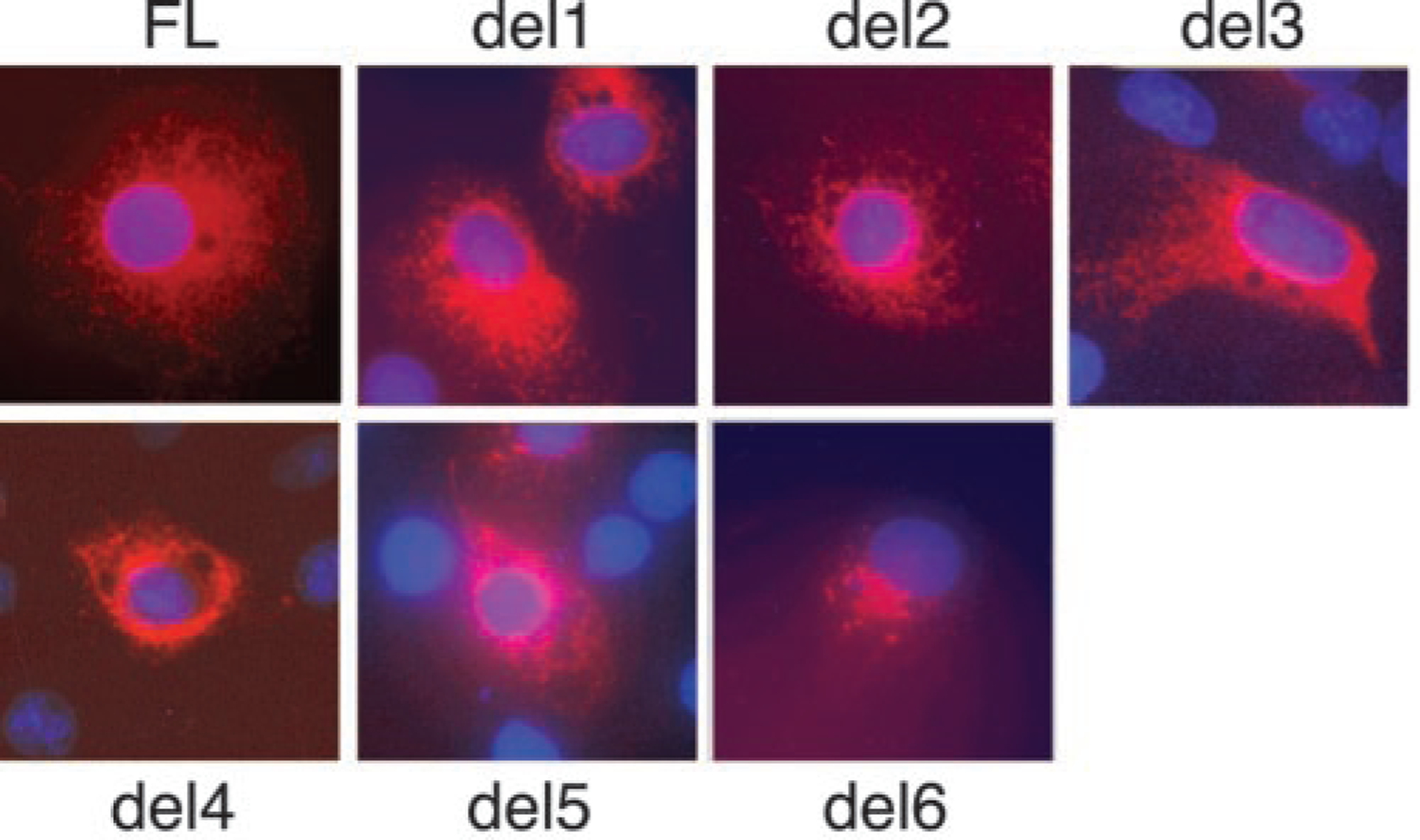
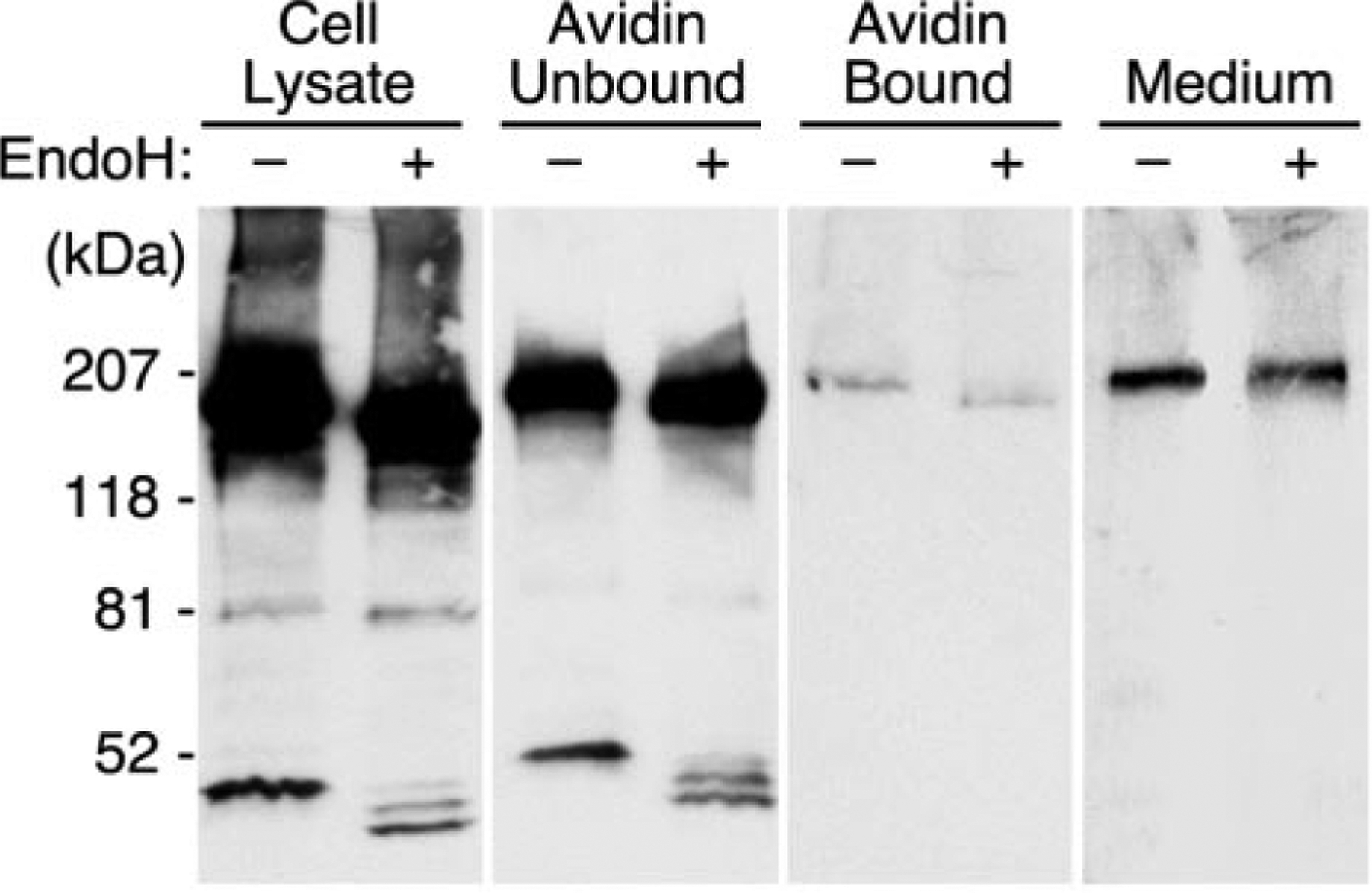
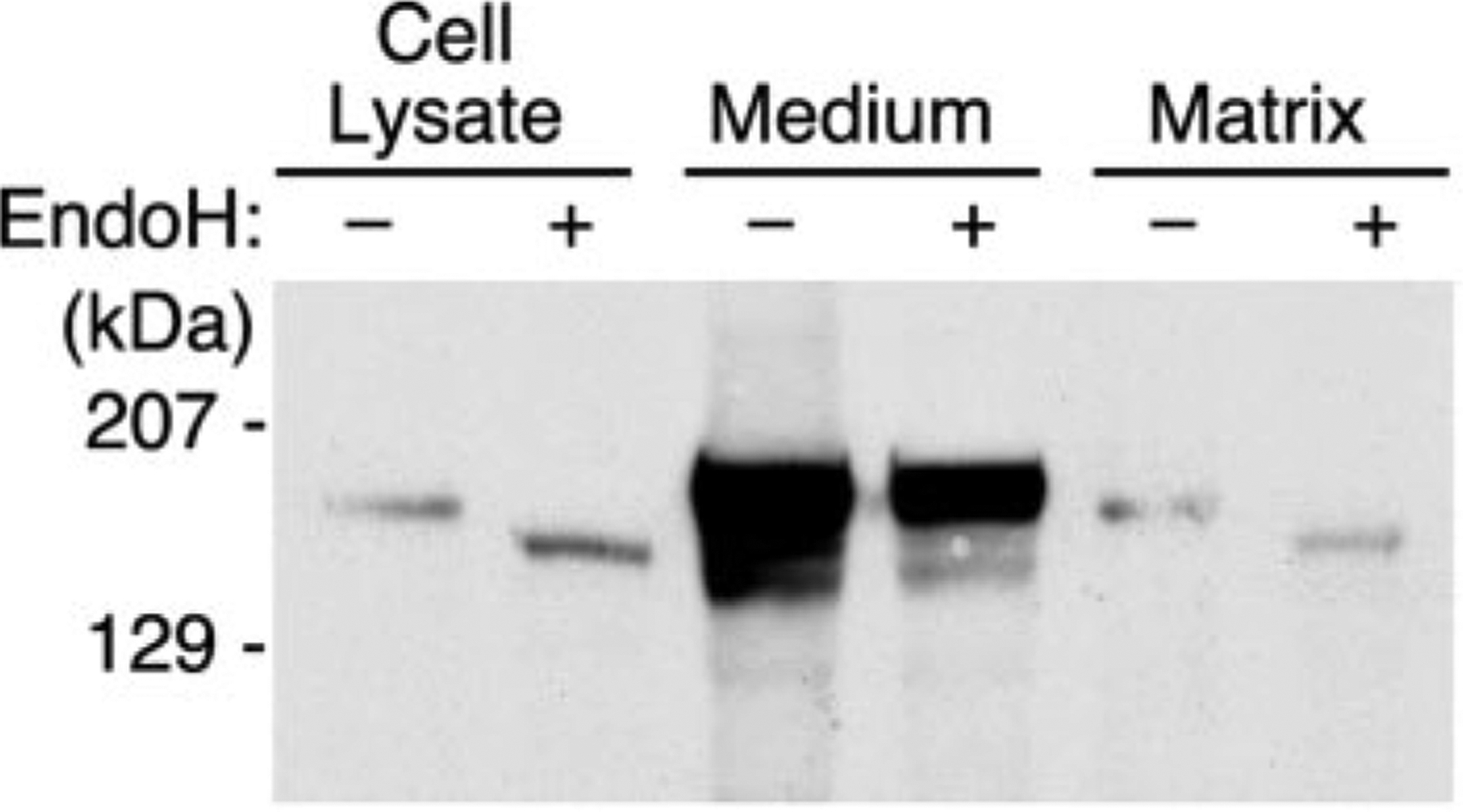
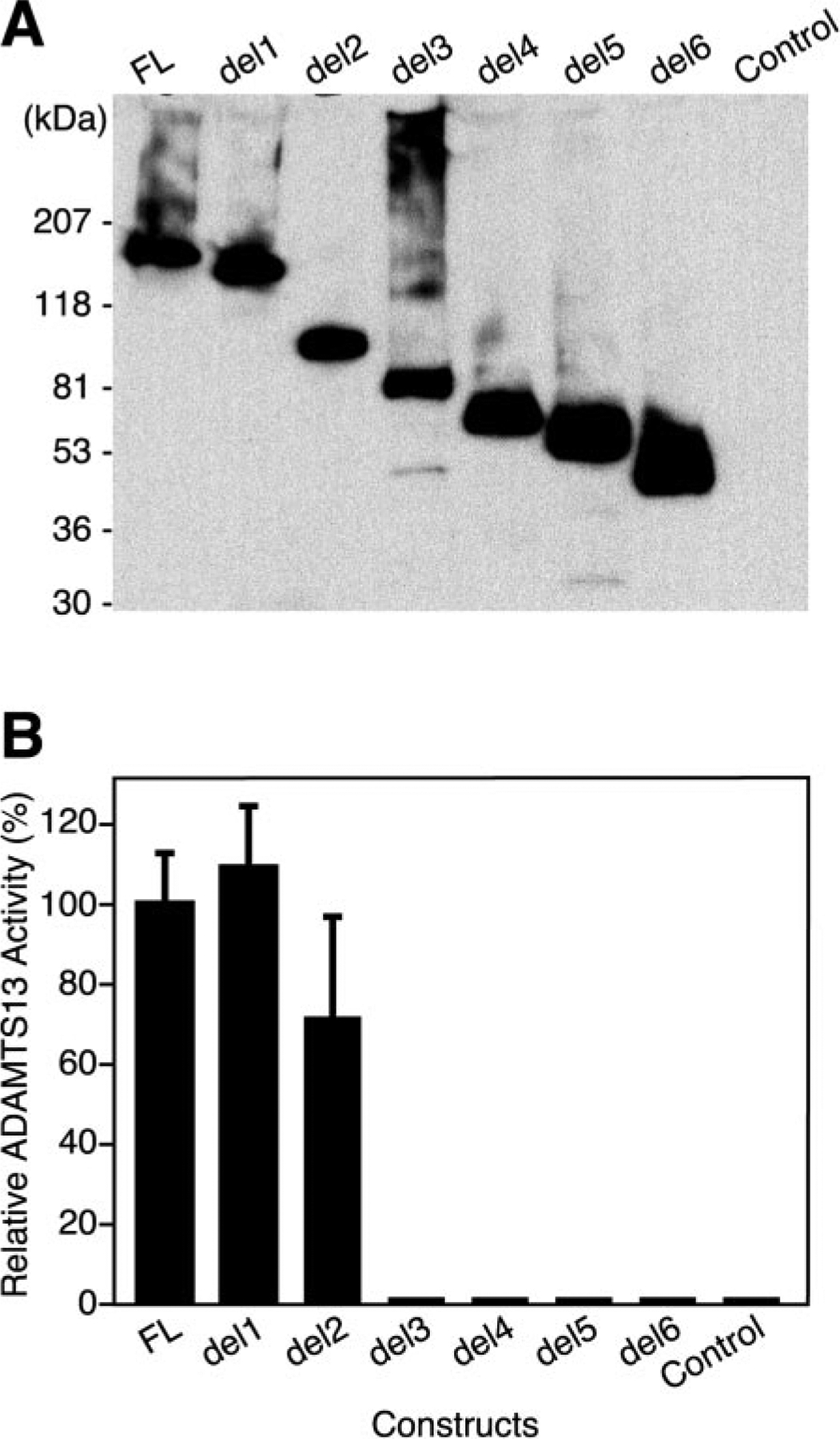
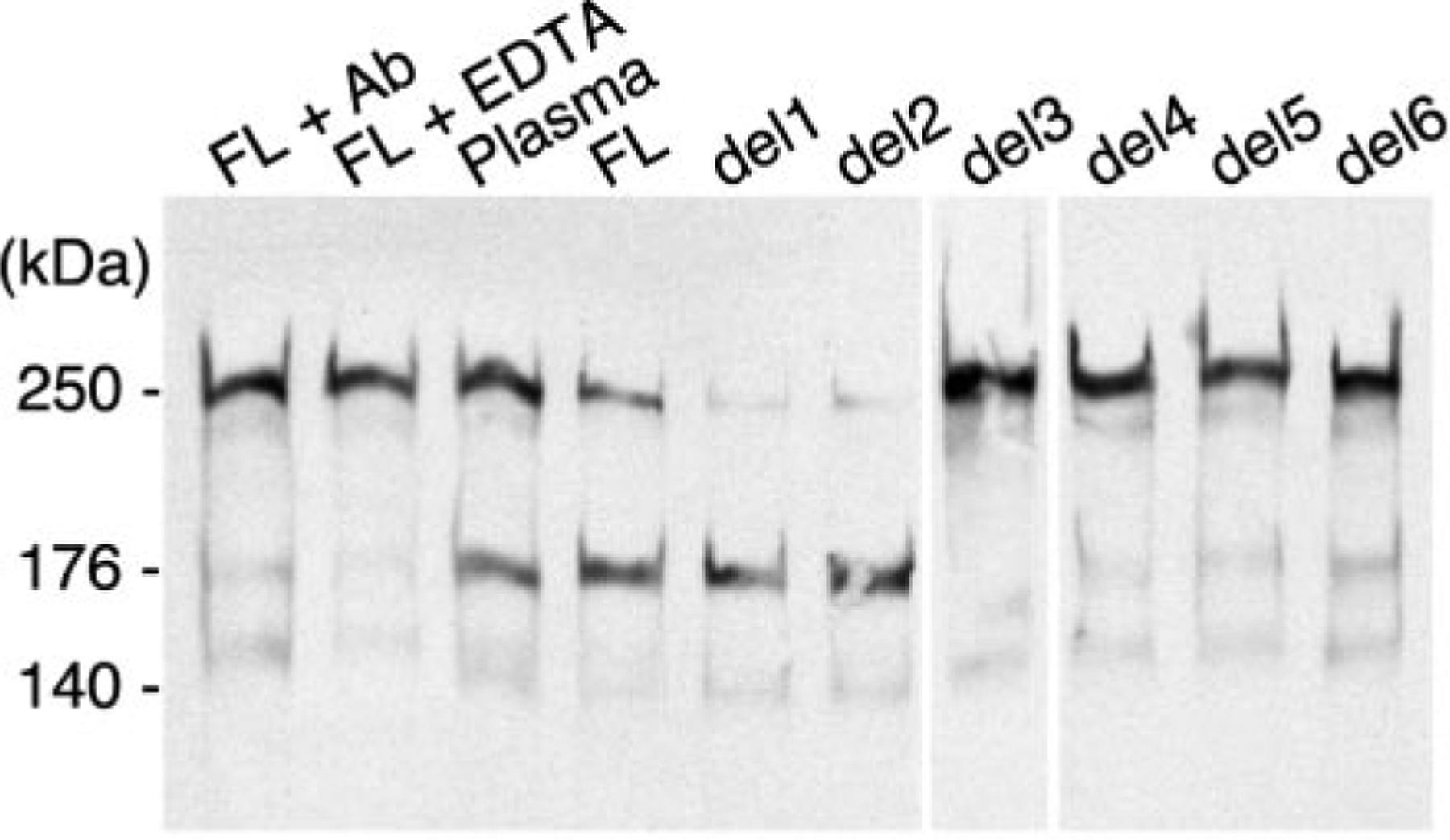
ADAMTS13 consists of a reprolysin-type metalloprotease domain followed by a disintegrin domain, a thrombospondin type 1 motif (TSP1), Cys-rich and spacer domains, seven more TSP1 motifs, and two CUB domains. ADAMTS13 limits platelet accumulation in microvascular thrombi by cleaving the Tyr1605-Met1606 bond in von Willebrand factor, and ADAMTS13 deficiency causes a lethal syndrome, thrombotic thrombocytopenic purpura. ADAMTS13 domains required for substrate recognition were localized by the characterization of recombinant deletion mutants. Constructs with C-terminal His6 and V5 epitopes were expressed by transient transfection of COS-7 cells or in a baculovirus system. No association with extracellular matrix or cell surface was detected for any ADAMTS13 variant by immunofluorescence microscopy or chemical modification. Both plasma and recombinant full-length ADAMTS13 cleaved von Willebrand factor subunits into two fragments of 176 kDa and 140 kDa. Recombinant ADAMTS13 was divalent metal ion-dependent and was inhibited by IgG from a patient with idiopathic thrombotic thrombocytopenic purpura. ADAMTS13 that was truncated after the metalloprotease domain, the disintegrin domain, the first TSP1 repeat, or the Cys-rich domain was not able to cleave von Willebrand factor, whereas addition of the spacer region restored protease activity. Therefore, the spacer region is necessary for normal ADAMTS13 activity toward von Willebrand factor, and the more C-terminal TSP1 and CUB domains are dispensable in vitro.
ADAMTS13由一个解聚素和金属蛋白酶结构域(reprolysin-type metalloprotease domain)、一个解聚素结构域(disintegrin domain)、一个血小板反应蛋白1型基序(thrombospondin type 1 motif,TSP1)、富含半胱氨酸和间隔结构域、另外七个TSP1基序以及两个CUB结构域组成。ADAMTS13通过切割血管性血友病因子(von Willebrand factor)中的Tyr1605-Met1606键来限制微血管血栓中血小板的聚集,而ADAMTS13缺乏会导致一种致命综合征——血栓性血小板减少性紫癜(thrombotic thrombocytopenic purpura)。通过对重组缺失突变体的表征确定了底物识别所需的ADAMTS13结构域。带有C末端His6和V5表位的构建体通过COS-7细胞的瞬时转染或在杆状病毒系统中表达。通过免疫荧光显微镜或化学修饰,未检测到任何ADAMTS13变体与细胞外基质或细胞表面有联系。血浆和重组全长ADAMTS13均可将血管性血友病因子亚基切割成176 kDa和140 kDa的两个片段。重组ADAMTS13依赖二价金属离子,并被一名特发性血栓性血小板减少性紫癜患者的IgG抑制。在金属蛋白酶结构域、解聚素结构域、第一个TSP1重复序列或富含半胱氨酸结构域之后被截断的ADAMTS13无法切割血管性血友病因子,而添加间隔区可恢复蛋白酶活性。因此,间隔区对于ADAMTS13对血管性血友病因子的正常活性是必需的,而更靠C末端的TSP1和CUB结构域在体外是可有可无的。