Brown Kimberly A, Roberts Richard L, Arteaga Carlos L, Law Brian K
Vanderbilt-Ingram Cancer Center, Vanderbilt University Medical Center, Nashville, TN, USA.
Breast Cancer Res. 2004;6(2):R130-9. doi: 10.1186/bcr762. Epub 2004 Feb 4.
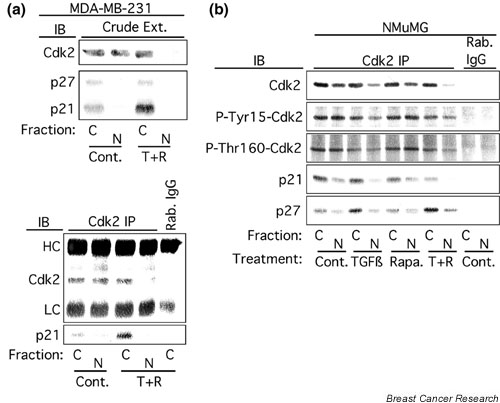
The transforming growth factor-beta (TGF-beta) signaling pathway functions to prevent tumorigenesis, and loss of sensitivity to TGF-beta-mediated cell cycle arrest is nearly ubiquitous among human cancers. Our previous studies demonstrated that rapamycin potentiates TGF-beta-induced cell cycle arrest in nontransformed epithelial cells and partially restores TGF-beta-induced growth arrest of some human cancer cell lines. Growth arrest correlated with increased binding of p21 and p27 to cyclin-dependent kinase-2 (Cdk2), and inhibition of Cdk2 kinase activity. However, it was unclear how TGF-beta caused increased binding of p21 and p27 to Cdk2.
Cell fractionation and immunofluorescence microscopy experiments were performed to examine the effect of TGF-beta on the intracellular localization of Cdk2, p21, and p27. Kinase assays were performed on cytoplasmic and nuclear extracts to determine how TGF-beta altered Cdk2 activity in both subcellular compartments.
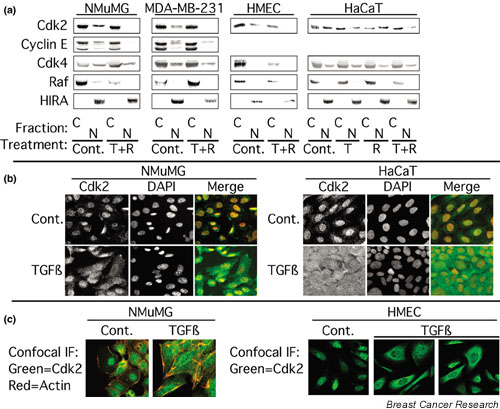
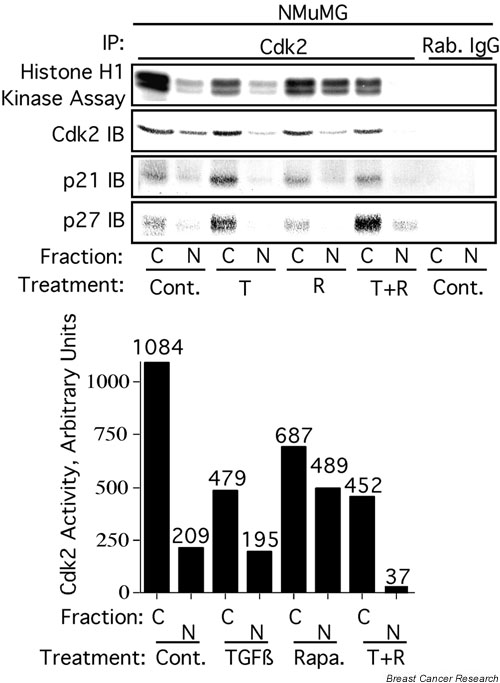
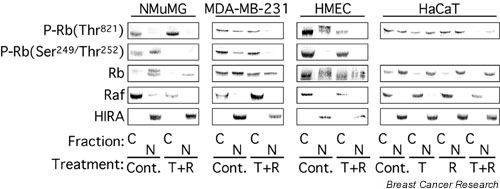
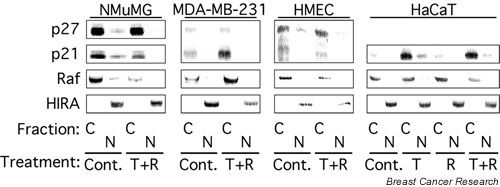
In breast epithelial cells treatment with TGF-beta induced a decrease in nuclear Cdk2 concentrations and relocalization of Cdk2 to the cytoplasm. Cdk2 relocalization to the cytoplasm correlated with dephosphorylation of nuclear retinoblastoma tumor suppressor protein and decreased nuclear Cdk2 activity. In these epithelial cell lines, p21 and p27 were localized primarily in the cytoplasm. Decreases in nuclear Cdk2 concentrations correlated with increased binding of Cdk2 to cytoplasmic p21 and p27.
Cooperative growth arrest induced by treatment with TGF-beta + rapamycin causes inhibition of nuclear Cdk2 activity through multiple mechanisms, including Cdk2 relocalization to the cytoplasm, increased p27 and p21 binding to Cdk2, and increased phosphorylation of nuclear Cdk2 on its inhibitory site, Tyr15.
转化生长因子-β(TGF-β)信号通路具有预防肿瘤发生的功能,而对TGF-β介导的细胞周期停滞的敏感性丧失在人类癌症中几乎普遍存在。我们之前的研究表明,雷帕霉素可增强TGF-β诱导的未转化上皮细胞的细胞周期停滞,并部分恢复TGF-β诱导的一些人类癌细胞系的生长停滞。生长停滞与p21和p27与细胞周期蛋白依赖性激酶-2(Cdk2)的结合增加以及Cdk2激酶活性的抑制相关。然而,尚不清楚TGF-β如何导致p21和p27与Cdk2的结合增加。
进行细胞分级分离和免疫荧光显微镜实验,以检查TGF-β对Cdk2、p21和p27细胞内定位的影响。对细胞质和细胞核提取物进行激酶测定,以确定TGF-β如何改变两个亚细胞区室中Cdk2的活性。
在用TGF-β处理的乳腺上皮细胞中,诱导核Cdk2浓度降低,并使Cdk2重新定位于细胞质。Cdk2重新定位于细胞质与核视网膜母细胞瘤肿瘤抑制蛋白的去磷酸化以及核Cdk2活性降低相关。在这些上皮细胞系中,p21和p27主要定位于细胞质。核Cdk2浓度的降低与Cdk2与细胞质p21和p27的结合增加相关。
TGF-β + 雷帕霉素联合处理诱导的协同生长停滞通过多种机制导致核Cdk2活性受到抑制,包括Cdk2重新定位于细胞质、p27和p21与Cdk2的结合增加以及核Cdk2在其抑制位点Tyr15上的磷酸化增加。