Harrington Laura S, Findlay Greg M, Gray Alex, Tolkacheva Tatiana, Wigfield Simon, Rebholz Heike, Barnett Jill, Leslie Nick R, Cheng Susan, Shepherd Peter R, Gout Ivan, Downes C Peter, Lamb Richard F
Cancer Research UK Centre for Cell and Molecular Biology, The Institute of Cancer Research, 237 Fulham Rd., London SW3 6JB, England, UK.
J Cell Biol. 2004 Jul 19;166(2):213-23. doi: 10.1083/jcb.200403069. Epub 2004 Jul 12.
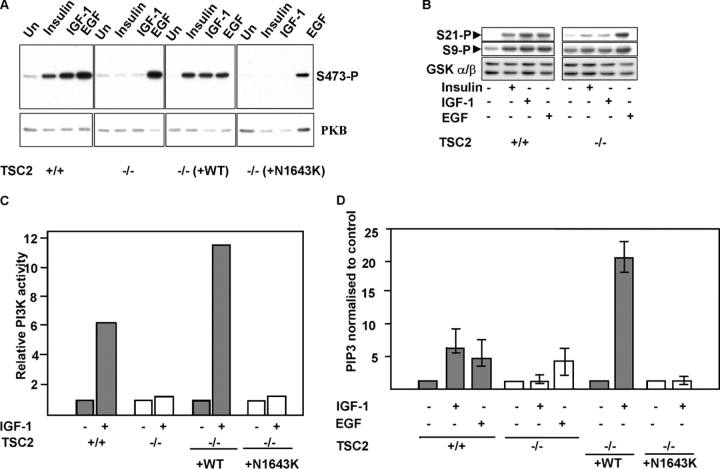
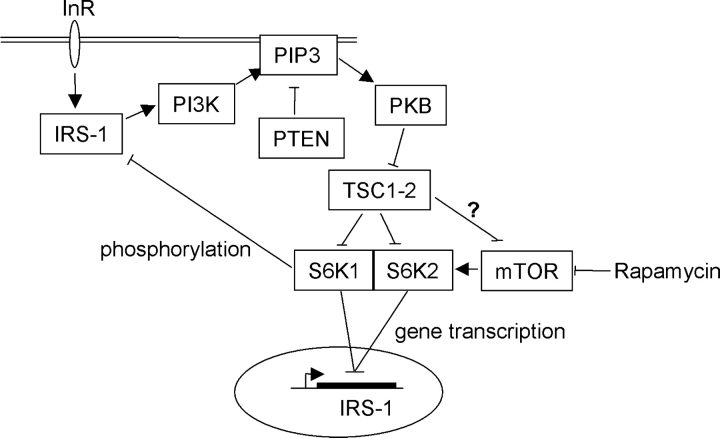
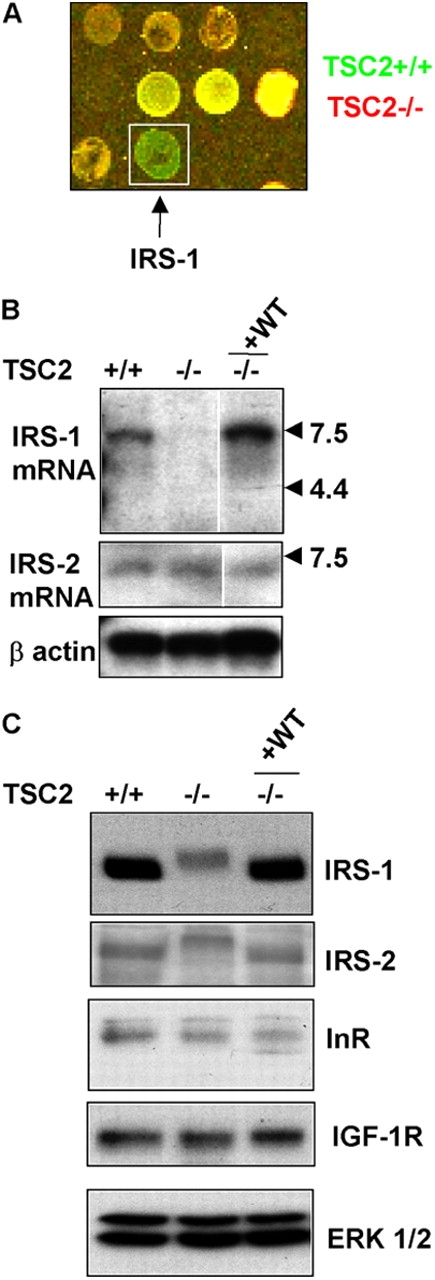
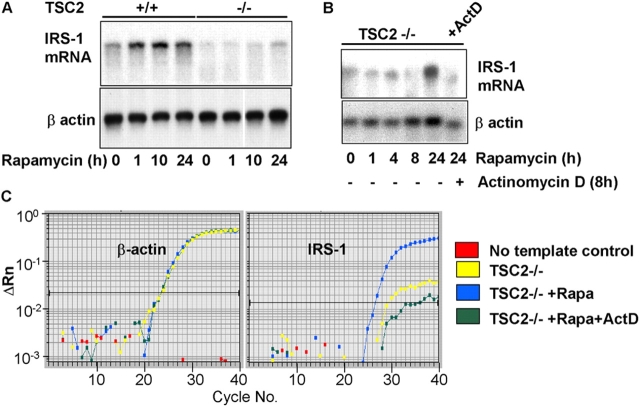
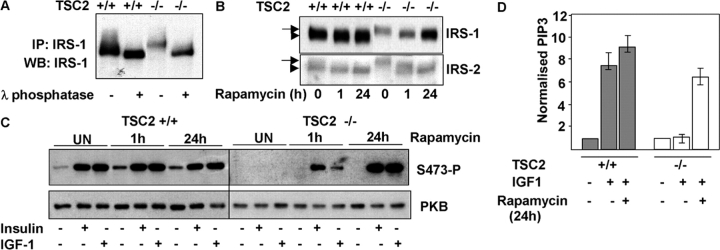
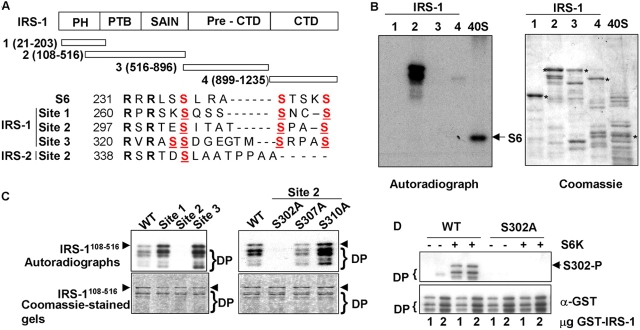
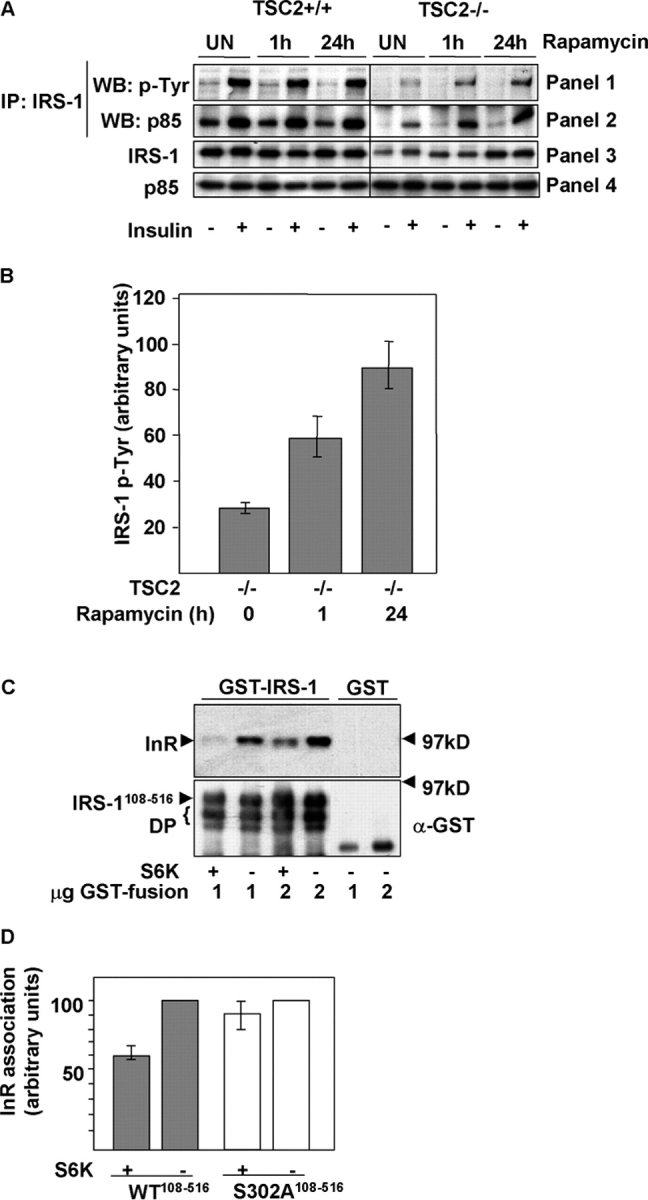
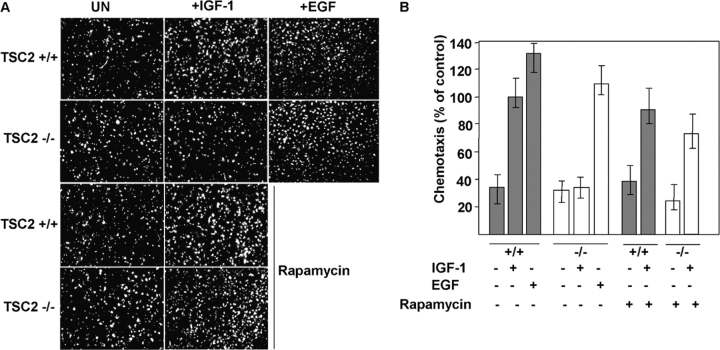
Insulin-like growth factors elicit many responses through activation of phosphoinositide 3-OH kinase (PI3K). The tuberous sclerosis complex (TSC1-2) suppresses cell growth by negatively regulating a protein kinase, p70S6K (S6K1), which generally requires PI3K signals for its activation. Here, we show that TSC1-2 is required for insulin signaling to PI3K. TSC1-2 maintains insulin signaling to PI3K by restraining the activity of S6K, which when activated inactivates insulin receptor substrate (IRS) function, via repression of IRS-1 gene expression and via direct phosphorylation of IRS-1. Our results argue that the low malignant potential of tumors arising from TSC1-2 dysfunction may be explained by the failure of TSC mutant cells to activate PI3K and its downstream effectors.
胰岛素样生长因子通过激活磷酸肌醇3 - 羟基激酶(PI3K)引发多种反应。结节性硬化复合物(TSC1 - 2)通过负向调节一种蛋白激酶p70S6K(S6K1)来抑制细胞生长,而p70S6K的激活通常需要PI3K信号。在此,我们表明TSC1 - 2是胰岛素向PI3K信号传导所必需的。TSC1 - 2通过抑制S6K的活性来维持胰岛素向PI3K的信号传导,当S6K被激活时,它会通过抑制IRS - 1基因表达和直接磷酸化IRS - 1来使胰岛素受体底物(IRS)功能失活。我们的结果表明,由TSC1 - 2功能障碍引起的肿瘤恶性潜能较低,这可能是由于TSC突变细胞无法激活PI3K及其下游效应器所致。