Majumdar Indraneel, Krishna S Sri, Grishin Nick V
Howard Hughes Medical Institute, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390, USA.
BMC Bioinformatics. 2005 Aug 11;6:202. doi: 10.1186/1471-2105-6-202.
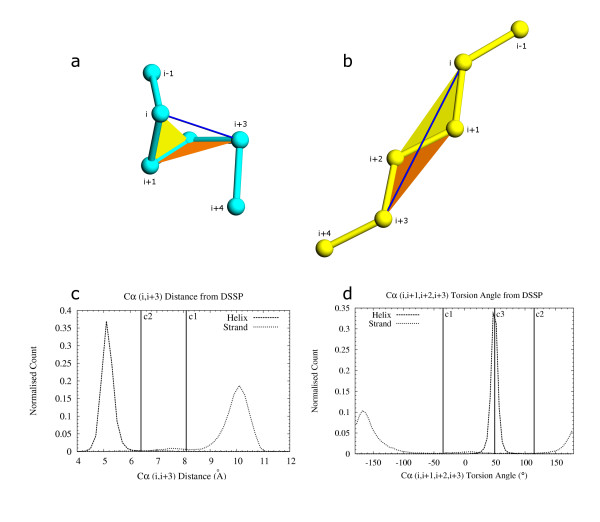
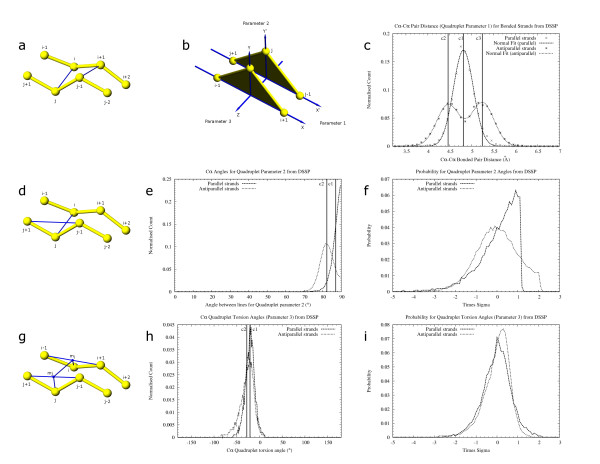
The majority of residues in protein structures are involved in the formation of alpha-helices and beta-strands. These distinctive secondary structure patterns can be used to represent a protein for visual inspection and in vector-based protein structure comparison. Success of such structural comparison methods depends crucially on the accurate identification and delineation of secondary structure elements.
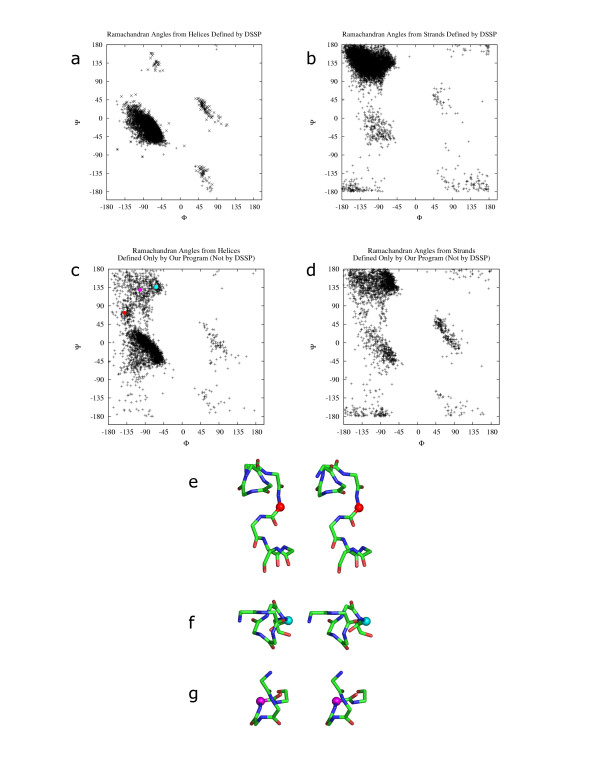
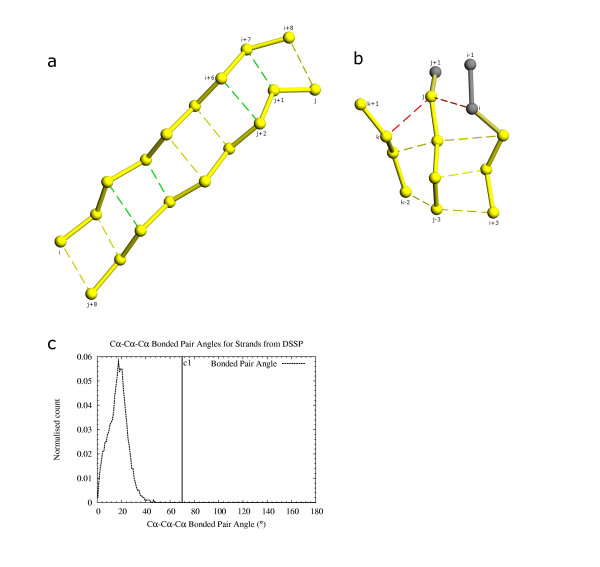
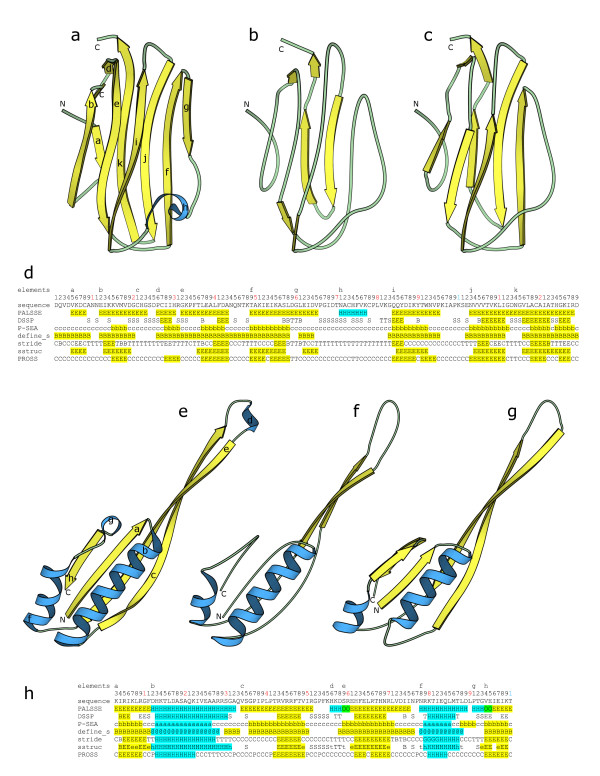
We have developed a method PALSSE (Predictive Assignment of Linear Secondary Structure Elements) that delineates secondary structure elements (SSEs) from protein Calpha coordinates and specifically addresses the requirements of vector-based protein similarity searches. Our program identifies two types of secondary structures: helix and beta-strand, typically those that can be well approximated by vectors. In contrast to traditional secondary structure algorithms, which identify a secondary structure state for every residue in a protein chain, our program attributes residues to linear SSEs. Consecutive elements may overlap, thus allowing residues located at the overlapping region to have more than one secondary structure type.
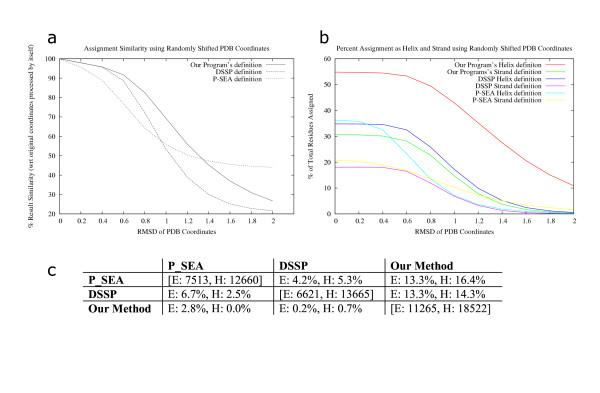
PALSSE is predictive in nature and can assign about 80% of the protein chain to SSEs as compared to 53% by DSSP and 57% by P-SEA. Such a generous assignment ensures almost every residue is part of an element and is used in structural comparisons. Our results are in agreement with human judgment and DSSP. The method is robust to coordinate errors and can be used to define SSEs even in poorly refined and low-resolution structures. The program and results are available at http://prodata.swmed.edu/palsse/.
蛋白质结构中的大多数残基参与α螺旋和β链的形成。这些独特的二级结构模式可用于表示蛋白质以便进行可视化检查以及基于向量的蛋白质结构比较。此类结构比较方法的成功关键取决于二级结构元件的准确识别和描绘。
我们开发了一种方法PALSSE(线性二级结构元件的预测性分配),该方法可从蛋白质的Cα坐标中描绘出二级结构元件(SSE),并特别满足基于向量的蛋白质相似性搜索的要求。我们的程序识别两种类型的二级结构:螺旋和β链,通常是那些可以用向量很好近似的结构。与传统的二级结构算法不同,传统算法会为蛋白质链中的每个残基识别一个二级结构状态,而我们的程序将残基归属于线性SSE。连续的元件可能会重叠,从而使位于重叠区域的残基具有不止一种二级结构类型。
PALSSE本质上具有预测性,与DSSP的53%和P-SEA的57%相比,它可以将约80%的蛋白质链分配给SSE。这样宽泛的分配确保了几乎每个残基都是一个元件的一部分,并用于结构比较。我们的结果与人类判断和DSSP一致。该方法对坐标误差具有鲁棒性,甚至可用于定义结构精修不佳和低分辨率结构中的SSE。该程序和结果可在http://prodata.swmed.edu/palsse/获取。