Hou Tingjun, Chen Ken, McLaughlin William A, Lu Benzhuo, Wang Wei
Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, California, United States of America.
PLoS Comput Biol. 2006 Jan;2(1):e1. doi: 10.1371/journal.pcbi.0020001. Epub 2006 Jan 27.
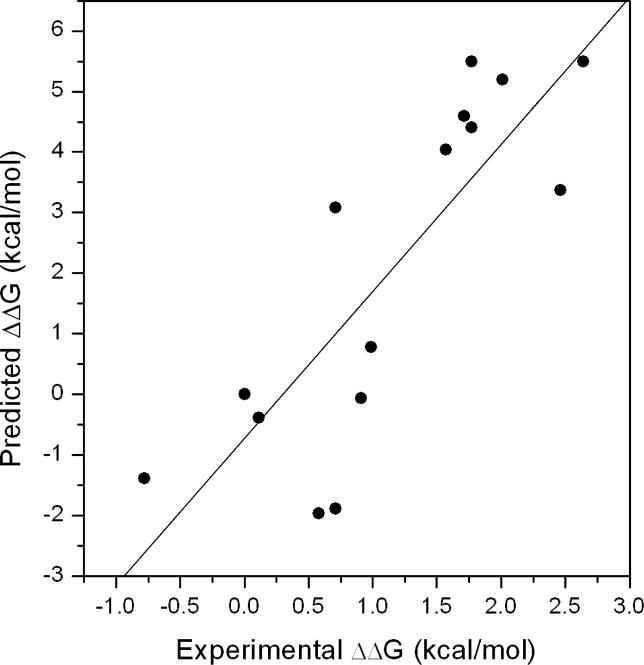
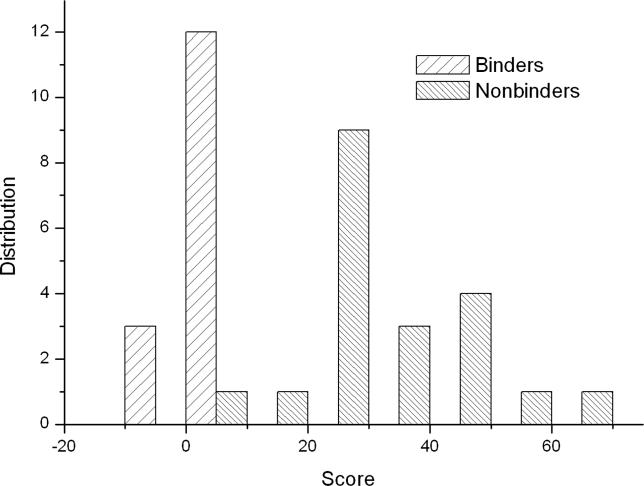
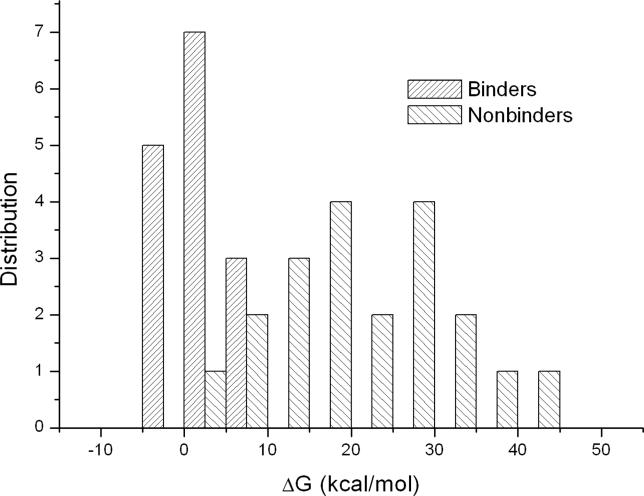
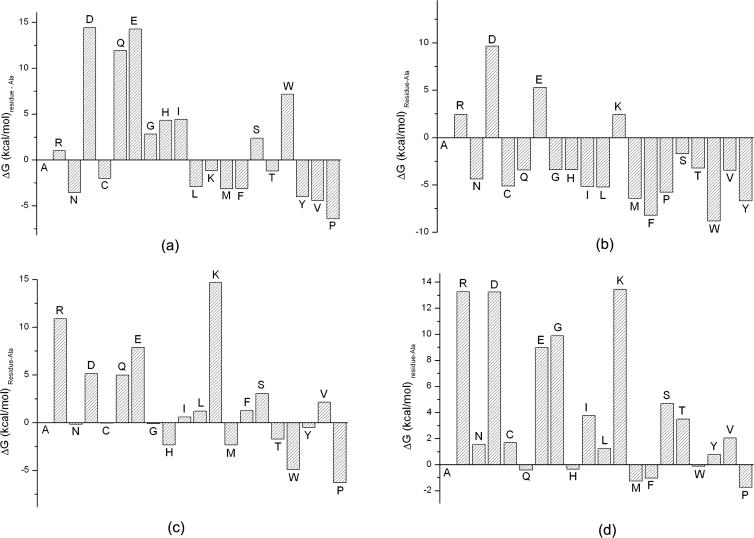
Protein-protein interactions, particularly weak and transient ones, are often mediated by peptide recognition domains, such as Src Homology 2 and 3 (SH2 and SH3) domains, which bind to specific sequence and structural motifs. It is important but challenging to determine the binding specificity of these domains accurately and to predict their physiological interacting partners. In this study, the interactions between 35 peptide ligands (15 binders and 20 non-binders) and the Abl SH3 domain were analyzed using molecular dynamics simulation and the Molecular Mechanics/Poisson-Boltzmann Solvent Area method. The calculated binding free energies correlated well with the rank order of the binding peptides and clearly distinguished binders from non-binders. Free energy component analysis revealed that the van der Waals interactions dictate the binding strength of peptides, whereas the binding specificity is determined by the electrostatic interaction and the polar contribution of desolvation. The binding motif of the Abl SH3 domain was then determined by a virtual mutagenesis method, which mutates the residue at each position of the template peptide relative to all other 19 amino acids and calculates the binding free energy difference between the template and the mutated peptides using the Molecular Mechanics/Poisson-Boltzmann Solvent Area method. A single position mutation free energy profile was thus established and used as a scoring matrix to search peptides recognized by the Abl SH3 domain in the human genome. Our approach successfully picked ten out of 13 experimentally determined binding partners of the Abl SH3 domain among the top 600 candidates from the 218,540 decapeptides with the PXXP motif in the SWISS-PROT database. We expect that this physical-principle based method can be applied to other protein domains as well.
蛋白质 - 蛋白质相互作用,尤其是弱相互作用和瞬时相互作用,通常由肽识别结构域介导,如Src同源结构域2和3(SH2和SH3),它们与特定的序列和结构基序结合。准确确定这些结构域的结合特异性并预测其生理相互作用伴侣既重要又具有挑战性。在本研究中,使用分子动力学模拟和分子力学/泊松 - 玻尔兹曼溶剂面积法分析了35种肽配体(15种结合剂和20种非结合剂)与Abl SH3结构域之间的相互作用。计算得到的结合自由能与结合肽的排序顺序相关性良好,并且能够清晰地区分结合剂和非结合剂。自由能成分分析表明,范德华相互作用决定了肽的结合强度,而结合特异性则由静电相互作用和去溶剂化的极性贡献决定。然后通过虚拟诱变方法确定Abl SH3结构域的结合基序,该方法将模板肽每个位置的残基相对于所有其他19种氨基酸进行突变,并使用分子力学/泊松 - 玻尔兹曼溶剂面积法计算模板肽和突变肽之间的结合自由能差异。由此建立了单位置突变自由能谱,并用作评分矩阵在人类基因组中搜索被Abl SH3结构域识别的肽。我们的方法在SWISS - PROT数据库中具有PXXP基序的218,540个十肽的前600个候选物中,成功地从13个实验确定的Abl SH3结构域结合伴侣中挑选出了10个。我们期望这种基于物理原理的方法也能应用于其他蛋白质结构域。