Center for Human Genetics, Duke University Medical Center, Durham, NC, USA.
BMC Genet. 2005 Dec 30;6 Suppl 1(Suppl 1):S148. doi: 10.1186/1471-2156-6-S1-S148.
Genomic screens generally employ a single-locus strategy for linkage analysis, but this may have low power in the presence of epistasis. Ordered subsets analysis (OSA) is a method for conditional linkage analysis using continuous covariates.
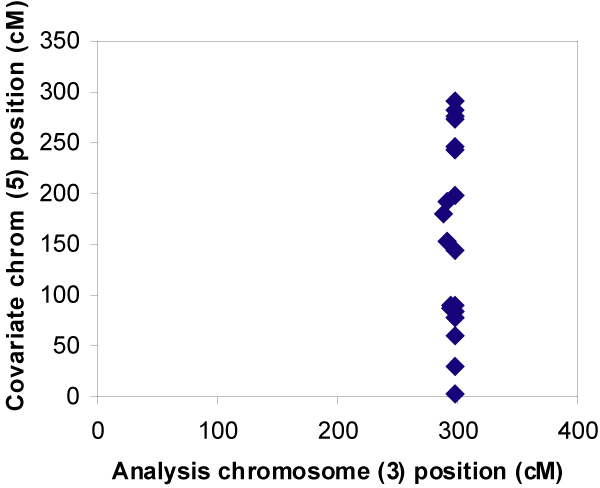
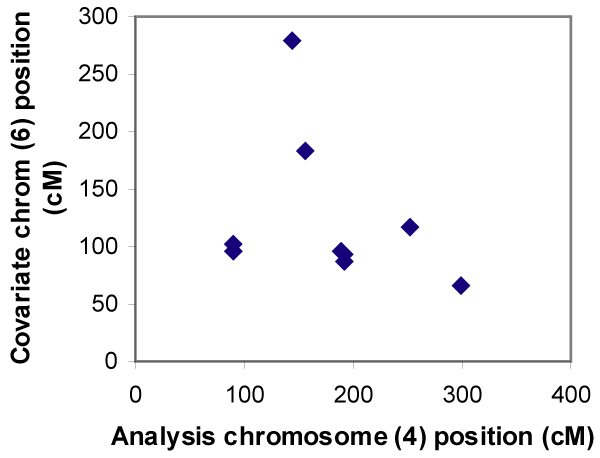
We used OSA to evaluate two-locus interactions in the simulated Genetic Analysis Workshop 14 dataset. We used all nuclear families ascertained by Aipotu, Karangar, and Danacaa. Using the single-nucleotide polymorphism map, multipoint affected-sibling-pair (ASP) linkage analysis was performed on all 100 replicates for each chromosome using SIBLINK. OSA was used to examine linkage on each chromosome using LOD scores at each 3-cM location on every other chromosome as covariates. Two methods were used to identify positive results: one searching across the entire covariate chromosome, the other conditioning on location of known disease loci.
Single-locus linkage analysis revealed very high LOD scores for disease loci D1 through D4, with mean LOD scores over 100 replicates ranging from 4.0 to 7.8. Although OSA did not obscure this linkage evidence, it did not detect the simulated interactions between any of the locus pairs. We found inflated type I error rates using the first OSA method, highlighting the need to correct for multiple comparisons. Therefore, using "null chromosome pairs" without simulated disease loci, we calculated a corrected alpha-level.
We were unable to detect two-locus interactions using OSA. This may have been due to lack of incorporation of phenotypic subgroups, or because linkage evidence as summarized by LOD scores performs poorly as an OSA covariate. We found inflated type I error rates, but were able to calculate a corrected alpha-level for future analyses employing this strategy to search for two-locus interactions.
基因组筛查通常采用单基因座策略进行连锁分析,但在存在上位性的情况下,这种策略的功效可能较低。有序子集分析(OSA)是一种使用连续协变量进行条件连锁分析的方法。
我们使用 OSA 评估了模拟遗传分析研讨会 14 数据集的两个基因座相互作用。我们使用了 Aipotu、Karangar 和 Danacaa 确定的所有核家庭。使用单核苷酸多态性图谱,使用 SIBLINK 对每条染色体上的所有 100 个重复进行了单点受影响的同胞对(ASP)连锁分析。使用 OSA,以每个其他染色体上每个 3cM 位置的 LOD 分数作为协变量,在每个染色体上进行连锁分析。使用两种方法来识别阳性结果:一种是在整个协变量染色体上进行搜索,另一种是在已知疾病基因座的位置上进行条件搜索。
单基因座连锁分析显示,疾病基因座 D1 至 D4 的 LOD 得分非常高,在 100 个重复中,平均 LOD 得分从 4.0 到 7.8 不等。尽管 OSA 没有掩盖这种连锁证据,但它没有检测到任何模拟基因座对之间的相互作用。我们发现使用第一种 OSA 方法会导致Ⅰ型错误率膨胀,这突出表明需要对多重比较进行校正。因此,使用没有模拟疾病基因座的“空”染色体对,我们计算了校正后的α水平。
我们无法使用 OSA 检测两个基因座的相互作用。这可能是由于缺乏对表型亚组的纳入,或者是由于 LOD 得分总结的连锁证据作为 OSA 协变量的表现不佳。我们发现了Ⅰ型错误率膨胀,但能够计算出校正后的α水平,以便将来使用这种策略搜索两个基因座的相互作用。