Epidemiology Unit, University of Manchester, Manchester, UK.
BMC Genet. 2005 Dec 30;6 Suppl 1(Suppl 1):S66. doi: 10.1186/1471-2156-6-S1-S66.
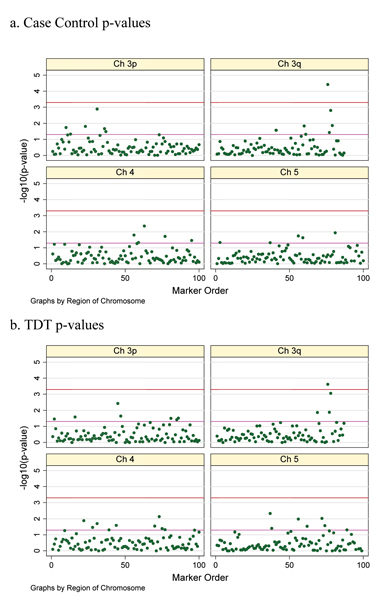
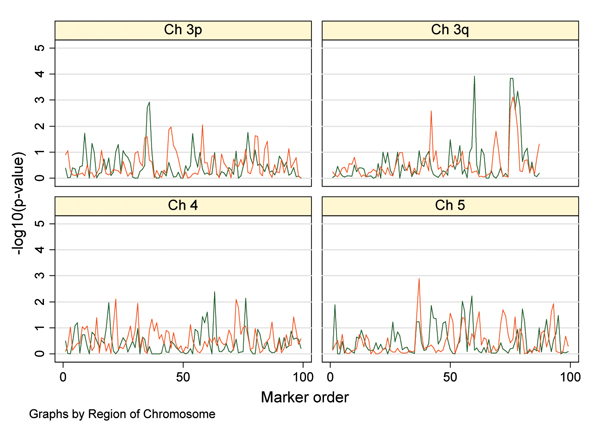
A common dilemma arising in linkage studies of complex genetic diseases is the selection of positive signals, their follow-up with association studies and discrimination between true and false positive results. Several strategies for overcoming these issues have been devised. Using the Genetic Analysis Workshop 14 simulated dataset, we aimed to apply different analytical approaches and evaluate their performance in discerning real associations. We considered a) haplotype analyses, b) different methods adjusting for multiple testing, c) replication in a second dataset, and d) exhaustive genotyping of all markers in a sufficiently powered, large sample group. We found that haplotype-based analyses did not substantially improve over single-point analysis, although this may reflect the low levels of linkage disequilibrium simulated in the datasets provided. Multiple testing correction methods were in general found to be over-conservative. Replication of nominally positive results in a second dataset appears to be less stringent, resulting in the follow-up of false positives. Performing a comprehensive assay of all markers in a large, well-powered dataset appears to be the most effective strategy for complex disease gene identification.
在复杂遗传疾病的连锁研究中,常会遇到一个难题,即如何选择阳性信号,如何对其进行关联研究,并区分真实阳性结果和假阳性结果。目前已经设计出了几种克服这些问题的策略。我们使用了遗传分析工作坊 14 模拟数据集,旨在应用不同的分析方法,并评估它们在辨别真实关联方面的性能。我们考虑了 a)单体型分析,b)用于多重检验调整的不同方法,c)在第二个数据集的复制,以及 d)在一个具有足够效力的大型样本组中对所有标记进行详尽的基因分型。我们发现,基于单体型的分析并没有比单点分析有实质性的改进,尽管这可能反映了所提供数据集模拟的低水平连锁不平衡。一般来说,多重检验校正方法被发现过于保守。在第二个数据集中复制名义上的阳性结果似乎不太严格,导致假阳性结果的后续跟踪。在一个大型、功能强大的数据集上全面检测所有标记似乎是识别复杂疾病基因的最有效策略。