Cancer Prevention Program, Fred Hutchinson Cancer Research Center, Seattle, WA, USA.
BMC Genet. 2005 Dec 30;6 Suppl 1(Suppl 1):S82. doi: 10.1186/1471-2156-6-S1-S82.
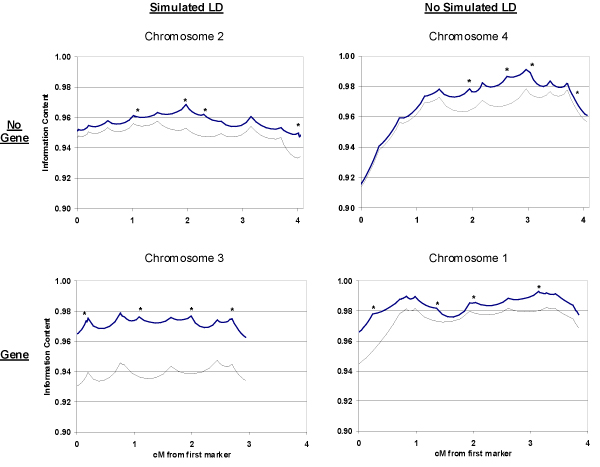
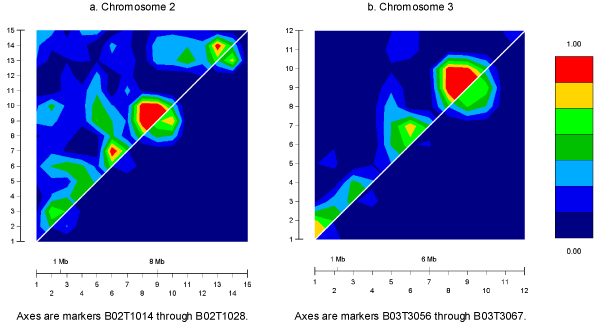
Current genome-wide linkage-mapping single-nucleotide polymorphism (SNP) panels with densities of 0.3 cM are likely to have increased intermarker linkage disequilibrium (LD) compared to 5-cM microsatellite panels. The resulting difference in haplotype frequencies versus that predicted may affect multipoint linkage analysis with ungenotyped founders; a common haplotype may be assumed to be rare, leading to inflation of identical-by-descent (IBD) allele-sharing estimates and evidence for linkage. Using data simulated for the Genetic Analysis Workshop 14, we assessed bias in allele-sharing measures and nonparametric linkage (NPL all) and Kong and Cox LOD (KC-LOD) scores in a targeted analysis of regions with and without LD and with and without genes. Using over 100 replicates, we found that if founders were not genotyped, multipoint IBD estimates and delta parameters were modestly inflated and NPL all and KC-LOD scores were biased upwards in the region with LD and no gene; rather than centering on the null, the mean NPL all and KC-LOD scores were 0.51 +/- 0.91 and 0.19 +/- 0.38, respectively. Reduction of LD by dropping markers reduced this upward bias. These trends were not seen in the non-LD region with no gene. In regions with genes (with and without LD), a slight loss in power with dropping markers was suggested. These results indicate that LD should be considered in dense scans; removal of markers in LD may reduce false-positive results although information may also be lost. Methods to address LD in a high-throughput manner are needed for efficient, robust genomic scans with dense SNPs.
目前,基因组范围内的单核苷酸多态性(SNP)连锁图谱密度为 0.3cM,与 5cM 微卫星图谱相比,可能具有更高的标记间连锁不平衡(LD)。这种单倍型频率的差异可能会影响未分型的起始者的多点连锁分析;常见的单倍型可能被认为是罕见的,从而导致同源等位基因共享估计值的膨胀和连锁的证据。我们使用 14 届遗传分析研讨会的数据进行模拟,评估了在 LD 存在和不存在的区域以及有和没有基因的区域进行靶向分析时,等位基因共享测量值和非参数连锁(NPL 全部)和 Kong 和 Cox LOD(KC-LOD)得分的偏差。使用超过 100 次重复,我们发现如果起始者没有进行基因分型,多点 IBD 估计值和 delta 参数会适度膨胀,并且在 LD 不存在基因的区域中,NPL 全部和 KC-LOD 得分会向上偏差;而不是以零为中心,平均 NPL 全部和 KC-LOD 得分分别为 0.51 +/- 0.91 和 0.19 +/- 0.38。通过删除标记物减少 LD 可以减少这种向上的偏差。在没有基因的非 LD 区域中,没有观察到这些趋势。在有基因的区域(有和没有 LD)中,标记物的删除表明存在轻微的功效损失。这些结果表明,在密集扫描中应考虑 LD;去除 LD 中的标记可能会减少假阳性结果,尽管信息也可能丢失。需要高效、稳健的基因组扫描方法来解决高密度 SNP 中的 LD 问题。