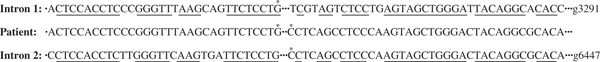Shabbeer Junaid, Yasuda Makiko, Benson Stacy D, Desnick Robert J
Department of Human Genetics, Mount Sinai School of Medicine, New York University, New York, NY 10029, USA.
Hum Genomics. 2006 Mar;2(5):297-309. doi: 10.1186/1479-7364-2-5-297.
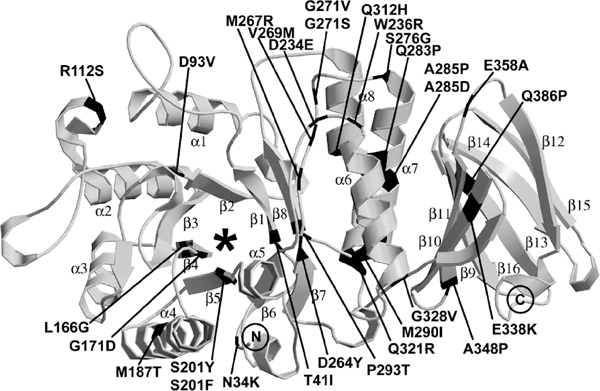
Fabry disease, an X-linked recessive inborn error of glycosphingolipid catabolism, results from the deficient activity of the lysosomal exoglycohydrolase, alpha-galactosidase A (EC 3.2.1.22; alpha-Gal A). The molecular lesions in the alpha-Gal A gene causing the classic phenotype of Fabry disease in 66 unrelated families were determined. In 49 families, 50 new mutations were identified, including: 29 missense mutations (N34K, T41I, D93V, R112S, L166G, G171D, M187T, S201Y, S201F, D234E, W236R, D264Y, M267R, V269M, G271S, G271V, S276G, Q283P, A285P, A285D, M290I, P293T, Q312H, Q321R, G328V, E338K, A348P, E358A, Q386P); nine nonsense mutations (C56X, E79X, K127X, Y151X, Y173X, L177X, W262X, Q306X, E338X); five splicing defects (IVS4-1G>A, IVS5-2A>G, IVS5+3A>G, IVS5+4A>G, IVS6-1G>C); four small deletions (18delA, 457delGAC, 567delG, 1096delACCAT); one small insertion (996insC); one 3.1 kilobase Alu-Alu deletion (which included exon 2); and one complex mutation (K374R, 1124delGAG). In 18 families, 17 previously reported mutations were identified, with R112C occurring in two families. In two classically affected families, affected males were identified with two mutations: one with two novel mutations, D264Y and V269M and the other with one novel (Q312H) and one previously reported (A143T) mutation. Transient expression of the individual mutations revealed that D264Y and Q312H were localised in the endoplasmic reticulum and had no detectable or markedly reduced activity, whereas V269M and A143T were localised in lysosomes and had approximately 10 per cent and approximately 35 per cent of expressed wild-type activity, respectively. Structural analyses based on the enzyme's three-dimensional structure predicted the effect of the 29 novel missense mutations on the mutant glycoprotein's structure. Of note, three novel mutations (approximately 10 per cent) were predicted not to significantly alter the glycoprotein's structure; however, they were disease causing. These studies further define the molecular heterogeneity of the alpha-Gal A mutations in classical Fabry disease, permit precise heterozygote detection and prenatal diagnosis, and provide insights into the structural alterations of the mutant enzymes that cause the classic phenotype.
法布里病是一种X连锁隐性糖鞘脂分解代谢先天性疾病,由溶酶体外切糖苷酶α-半乳糖苷酶A(EC 3.2.1.22;α-Gal A)活性缺乏所致。我们确定了66个无关家族中导致法布里病经典表型的α-Gal A基因的分子病变。在49个家族中,鉴定出50个新突变,包括:29个错义突变(N34K、T41I、D93V、R112S、L166G、G171D、M187T、S201Y、S201F、D234E、W236R、D264Y、M267R、V269M、G271S、G271V、S276G、Q283P、A285P、A285D、M290I、P293T、Q312H、Q321R、G328V、E338K、A348P、E358A、Q386P);9个无义突变(C56X、E79X、K127X、Y151X、Y173X、L177X、W262X、Q306X、E338X);5个剪接缺陷(IVS4-1G>A、IVS5-2A>G、IVS5+3A>G、IVS5+4A>G、IVS6-1G>C);4个小缺失(18delA、457delGAC、567delG、1096delACCAT);1个小插入(996insC);1个3.1千碱基的Alu-Alu缺失(包括外显子2);以及1个复合突变(K374R,1124delGAG)。在18个家族中,鉴定出17个先前报道的突变,其中R112C在两个家族中出现。在两个典型受累家族中,受累男性被鉴定出有两个突变:一个有两个新突变,D264Y和V269M,另一个有一个新突变(Q312H)和一个先前报道的突变(A143T)。单个突变的瞬时表达显示,D264Y和Q312H定位于内质网,且活性检测不到或显著降低,而V269M和A143T定位于溶酶体,其表达的野生型活性分别约为10%和约35%。基于该酶三维结构的结构分析预测了29个新错义突变对突变糖蛋白结构的影响。值得注意的是,三个新突变(约10%)预计不会显著改变糖蛋白的结构;然而,它们是致病的。这些研究进一步明确了经典法布里病中α-Gal A突变的分子异质性,允许进行精确的杂合子检测和产前诊断,并为导致经典表型的突变酶的结构改变提供了见解。