Gamundi María José, Hernan Imma, Martínez-Gimeno María, Maseras Miquel, García-Sandoval Blanca, Ayuso Carmen, Antiñolo Guillermo, Baiget Montserrat, Carballo Miguel
Servicio de Laboratorio, Hospital de Terrassa, Ctra, Torrebonica s/n 08227 Terrassa, Barcelona, España.
BMC Med Genet. 2006 Apr 5;7:35. doi: 10.1186/1471-2350-7-35.
Retinitis pigmentosa (RP), a clinically and genetically heterogeneous group of retinal degeneration disorders affecting the photoreceptor cells, is one of the leading causes of genetic blindness. Mutations in the photoreceptor-specific gene RP1 account for 3-10% of cases of autosomal dominant RP (adRP). Most of these mutations are clustered in a 500 bp region of exon 4 of RP1.
Denaturing gradient gel electrophoresis (DGGE) analysis and direct genomic sequencing were used to evaluate the 5' coding region of exon 4 of the RP1 gene for mutations in 150 unrelated index adRP patients. Ophthalmic and electrophysiological examination of RP patients and relatives according to pre-existing protocols were carried out.
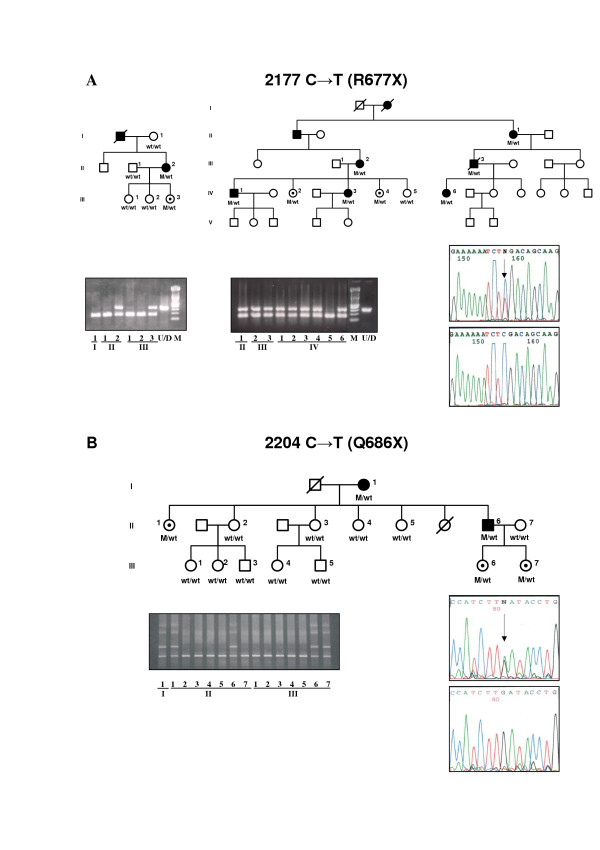
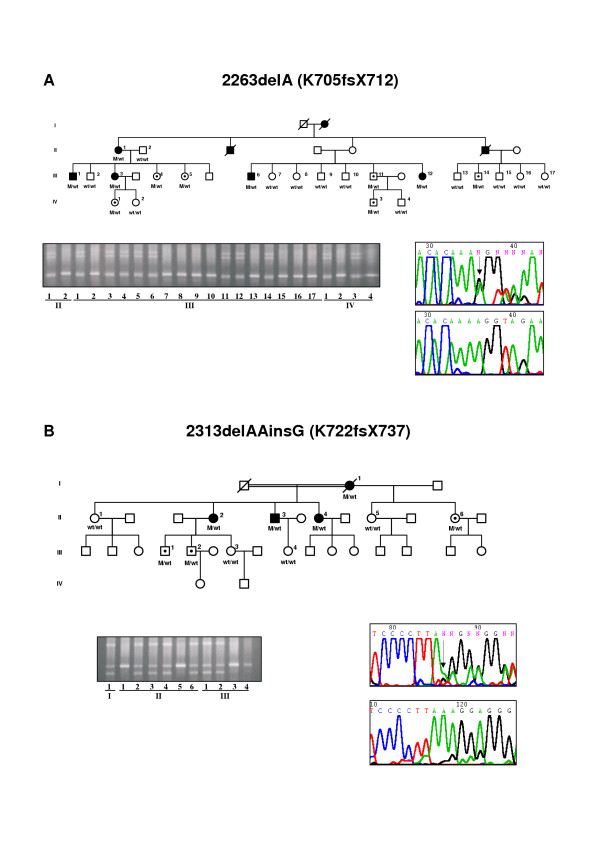
Three novel disease-causing mutations in RP1 were detected: Q686X, K705fsX712 and K722fsX737, predicting truncated proteins. One novel missense mutation, Thr752Met, was detected in one family but the mutation does not co-segregate in the family, thereby excluding this amino acid variation in the protein as a cause of the disease. We found the Arg677Ter mutation, previously reported in other populations, in two independent families, confirming that this mutation is also present in a Spanish population.
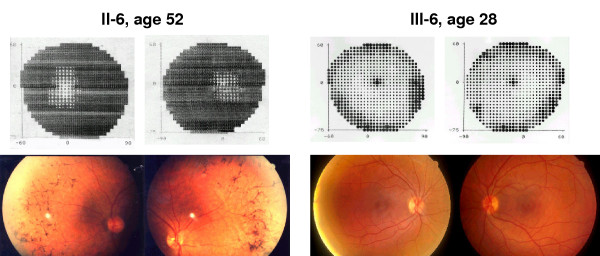
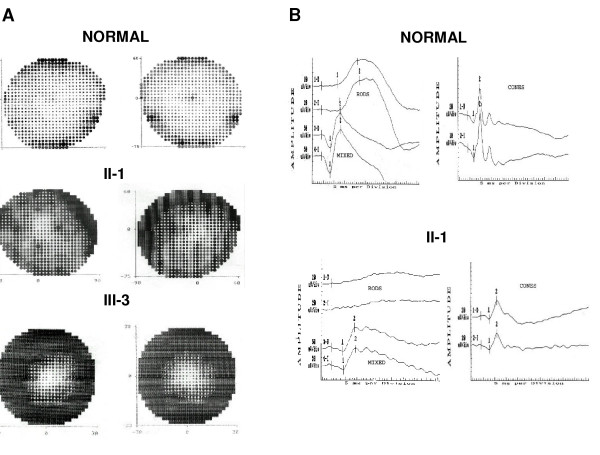
Most of the mutations reported in the RP1 gene associated with adRP are expected to encode mutant truncated proteins that are approximately one third or half of the size of wild type protein. Patients with mutations in RP1 showed mild RP with variability in phenotype severity. We also observed several cases of non-penetrant mutations.
视网膜色素变性(RP)是一组临床和遗传异质性的视网膜退行性疾病,影响光感受器细胞,是遗传性失明的主要原因之一。光感受器特异性基因RP1的突变占常染色体显性RP(adRP)病例的3%-10%。这些突变大多聚集在RP1外显子4的500bp区域。
采用变性梯度凝胶电泳(DGGE)分析和直接基因组测序,评估150例无关的adRP索引患者RP1基因外显子4的5'编码区是否存在突变。根据现有方案对RP患者及其亲属进行眼科和电生理检查。
在RP1中检测到3个新的致病突变:Q686X、K705fsX712和K722fsX737,预测会产生截短蛋白。在一个家族中检测到一个新的错义突变Thr752Met,但该突变在家族中不共分离,因此排除该蛋白质中的氨基酸变异是疾病的原因。我们在两个独立的家族中发现了先前在其他人群中报道过的Arg677Ter突变,证实该突变也存在于西班牙人群中。
与adRP相关的RP1基因中报道的大多数突变预计会编码大小约为野生型蛋白三分之一或一半的突变截短蛋白。RP1基因突变的患者表现为轻度RP,表型严重程度存在差异。我们还观察到几例非穿透性突变的病例。