Gilbert M Thomas P, Binladen Jonas, Miller Webb, Wiuf Carsten, Willerslev Eske, Poinar Hendrik, Carlson John E, Leebens-Mack James H, Schuster Stephan C
Center for Ancient Genetics, Niels Bohr Institute and Biological Institutes, The University of Copenhagen, Juliane Maries vej 30, DK-2100 Copenhagen Ø, Denmark.
Nucleic Acids Res. 2007;35(1):1-10. doi: 10.1093/nar/gkl483. Epub 2006 Aug 18.
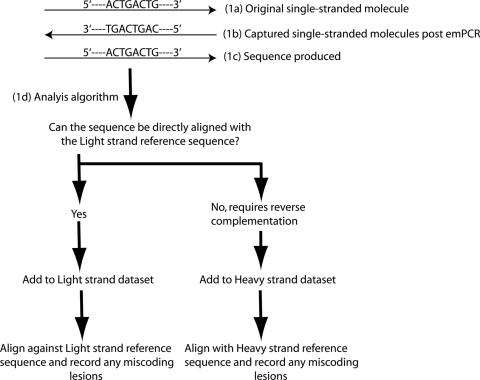
Although ancient DNA (aDNA) miscoding lesions have been studied since the earliest days of the field, their nature remains a source of debate. A variety of conflicting hypotheses exist about which miscoding lesions constitute true aDNA damage as opposed to PCR polymerase amplification error. Furthermore, considerable disagreement and speculation exists on which specific damage events underlie observed miscoding lesions. The root of the problem is that it has previously been difficult to assemble sufficient data to test the hypotheses, and near-impossible to accurately determine the specific strand of origin of observed damage events. With the advent of emulsion-based clonal amplification (emPCR) and the sequencing-by-synthesis technology this has changed. In this paper we demonstrate how data produced on the Roche GS20 genome sequencer can determine miscoding lesion strands of origin, and subsequently be interpreted to enable characterization of the aDNA damage behind the observed phenotypes. Through comparative analyses on 390,965 bp of modern chloroplast and 131,474 bp of ancient woolly mammoth GS20 sequence data we conclusively demonstrate that in this sample at least, a permafrost preserved specimen, Type 2 (cytosine-->thymine/guanine-->adenine) miscoding lesions represent the overwhelming majority of damage-derived miscoding lesions. Additionally, we show that an as yet unidentified guanine-->adenine analogue modification, not the conventionally argued cytosine-->uracil deamination, underpins a significant proportion of Type 2 damage. How widespread these implications are for aDNA will become apparent as future studies analyse data recovered from a wider range of substrates.
尽管自该领域早期以来就对古DNA(aDNA)错配损伤进行了研究,但其本质仍是一个争论的焦点。关于哪些错配损伤构成真正的aDNA损伤,而不是PCR聚合酶扩增错误,存在各种相互矛盾的假设。此外,对于观察到的错配损伤背后的具体损伤事件,也存在相当大的分歧和猜测。问题的根源在于,以前很难收集到足够的数据来检验这些假设,而且几乎不可能准确确定观察到的损伤事件的特定起源链。随着基于乳液的克隆扩增(emPCR)和合成测序技术的出现,这种情况已经改变。在本文中,我们展示了罗氏GS20基因组测序仪产生的数据如何确定错配损伤的起源链,随后对其进行解释,以表征观察到的表型背后的aDNA损伤。通过对390,965 bp的现代叶绿体和131,474 bp的古代猛犸象GS20序列数据进行比较分析,我们最终证明,至少在这个样本(一个永久冻土保存的标本)中,2型(胞嘧啶→胸腺嘧啶/鸟嘌呤→腺嘌呤)错配损伤占损伤衍生错配损伤的绝大多数。此外,我们表明,一种尚未确定的鸟嘌呤→腺嘌呤类似物修饰,而非传统认为的胞嘧啶→尿嘧啶脱氨基作用,是相当一部分2型损伤的基础。随着未来研究分析从更广泛的底物中回收的数据,这些影响对aDNA的广泛程度将变得明显。