Peng Junping, Zhang Xiaobing, Yang Jian, Wang Jing, Yang E, Bin Wen, Wei Candong, Sun Meisheng, Jin Qi
State Key Laboratory for Molecular Virology and Genetic Engineering, Chinese Center for Disease Control and Prevention, Beijing 100176, China.
BMC Genomics. 2006 Aug 29;7:218. doi: 10.1186/1471-2164-7-218.
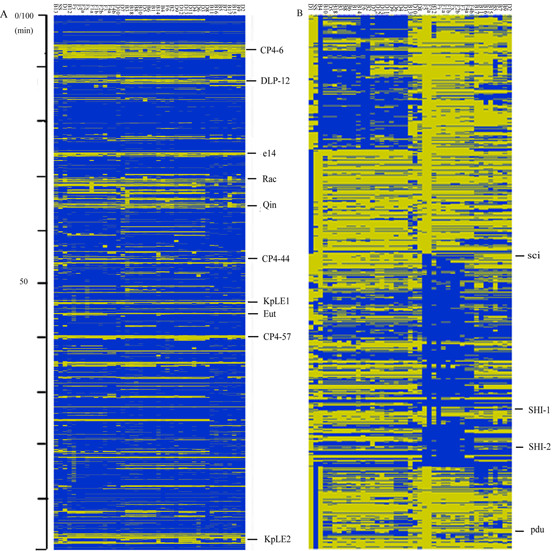
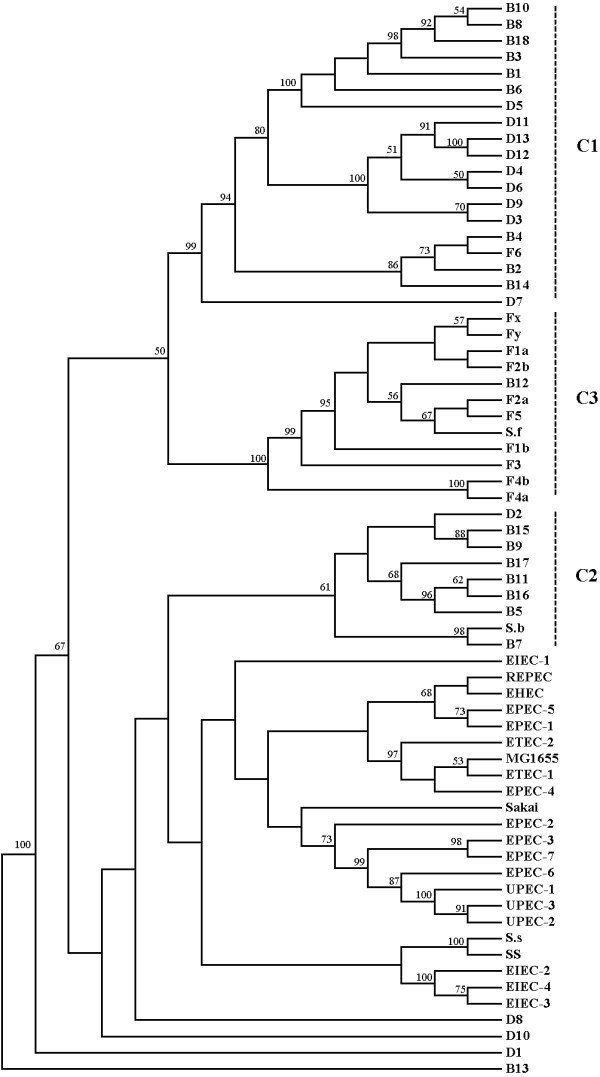
Compelling evidence indicates that Shigella species, the etiologic agents of bacillary dysentery, as well as enteroinvasive Escherichia coli, are derived from multiple origins of Escherichia coli and form a single pathovar. To further understand the genome diversity and virulence evolution of Shigella, comparative genomic hybridization microarray analysis was employed to compare the gene content of E. coli K-12 with those of 43 Shigella strains from all lineages.
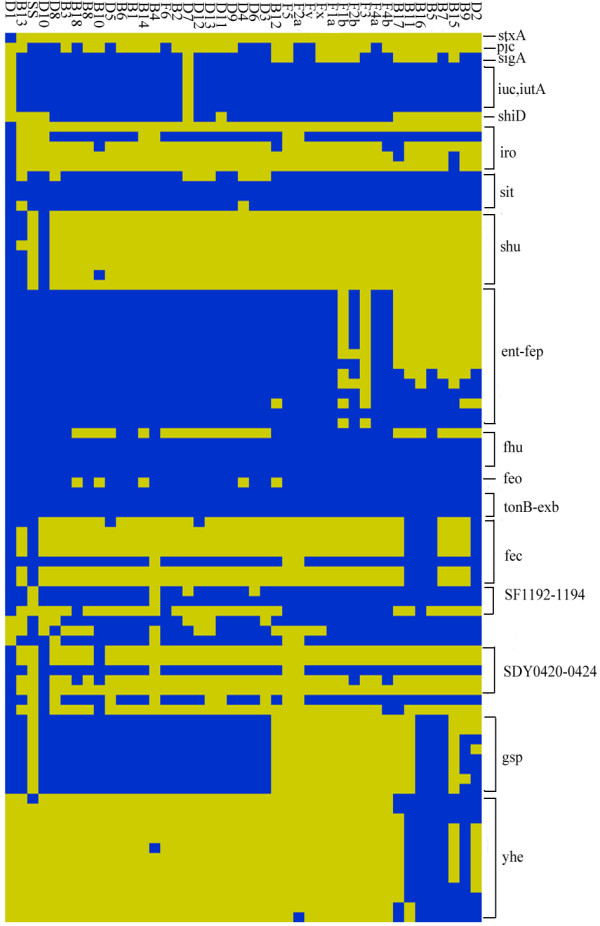
For the 43 strains subjected to CGH microarray analyses, the common backbone of the Shigella genome was estimated to contain more than 1,900 open reading frames (ORFs), with a mean number of 726 undetectable ORFs. The mosaic distribution of absent regions indicated that insertions and/or deletions have led to the highly diversified genomes of pathogenic strains.
These results support the hypothesis that by gain and loss of functions, Shigella species became successful human pathogens through convergent evolution from diverse genomic backgrounds. Moreover, we also found many specific differences between different lineages, providing a window into understanding bacterial speciation and taxonomic relationships.
有力证据表明,志贺氏菌属(细菌性痢疾的病原体)以及侵袭性大肠杆菌均源自大肠杆菌的多个起源,并形成一个单一的致病型。为了进一步了解志贺氏菌的基因组多样性和毒力进化,采用比较基因组杂交微阵列分析来比较大肠杆菌K-12与来自所有谱系的43株志贺氏菌菌株的基因含量。
对于接受CGH微阵列分析的43株菌株,志贺氏菌基因组的共同主干估计包含超过1900个开放阅读框(ORF),平均有726个无法检测到的ORF。缺失区域的镶嵌分布表明插入和/或缺失导致了致病菌株基因组的高度多样化。
这些结果支持这样的假设,即通过功能的获得和丧失,志贺氏菌属通过从不同基因组背景的趋同进化成为成功的人类病原体。此外,我们还发现了不同谱系之间的许多特定差异,为理解细菌物种形成和分类关系提供了一个窗口。