De Marco L, Mazzucato M, Fabris F, De Roia D, Coser P, Girolami A, Vicente V, Ruggeri Z M
Centro Trasfusionale e Chimica Clinica, C.R.O. Aviano, Pordenone, Italy.
J Clin Invest. 1990 Jul;86(1):25-31. doi: 10.1172/JCI114692.
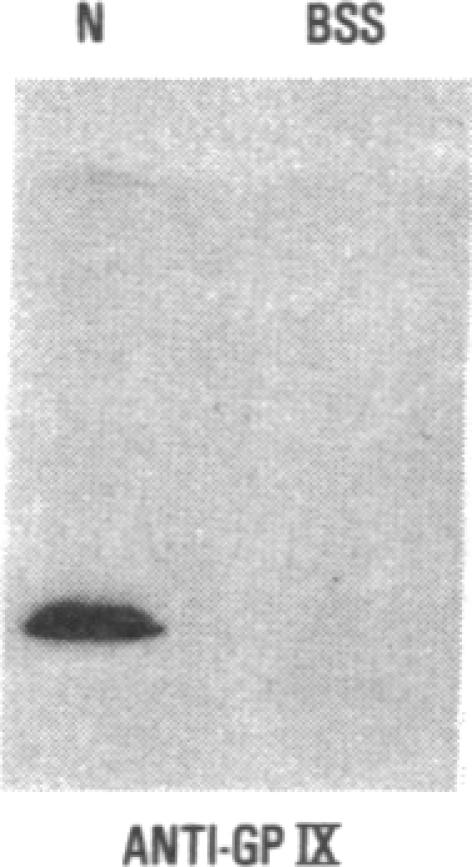
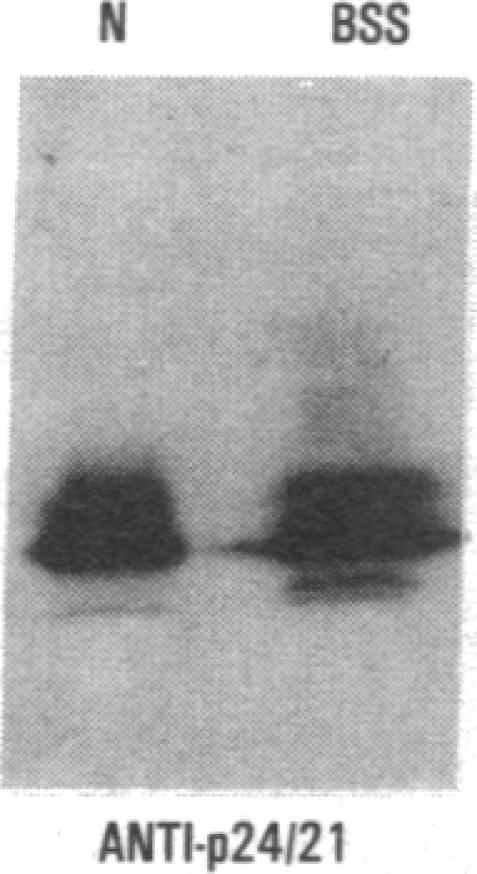
We have studied a patient with a congenital bleeding disorder and phenotypic manifestations typical of Bernard-Soulier syndrome, including giant platelets with absent ristocetin-induced von Willebrand factor binding. Two monoclonal antibodies reacting with distinct epitopes in the amino-terminal domain of the alpha-chain of glycoprotein (GP) Ib were used to estimate the number of GP Ib molecules on the platelet membrane. In the patient, binding of one antibody (LJ-Ib10) was approximately 50% of normal, while binding of the other (LJ-Ib1) was absent. Binding of both antibodies was reduced to approximately 50% of normal in the mother and one sister of the propositus, and their platelets exhibited approximately 70% of normal von Willebrand factor binding. Immunoblotting studies confirmed the presence of GP Ib alpha, as well as GP IX, in patient platelets. Antibody LJ-Ib10, but not LJ-Ib1, could immunoprecipitate the patient's GP Ib alpha from surface-labeled proteins. Thus, platelets from the propositus contained a structurally and functionally altered GP Ib-IX complex lacking a specific antibody epitope and the ability to bind von Willebrand factor. In contrast, the binding of human alpha-thrombin to the patient's platelets was normal, and three classes of binding sites with high, intermediate, and low affinity could be detected. These studies define a distinct variant form of Bernard-Soulier syndrome and provide evidence, based on a naturally occurring mutant molecule, that the amino-terminal region of GP Ib alpha contains a von Willebrand factor-binding domain distinct from the high affinity thrombin-binding site. Use of different monoclonal antibodies with distinct epitope specificities appears to be essential for a correct identification of variant Bernard-Soulier syndrome.
我们研究了一名患有先天性出血性疾病且具有典型伯纳德-索利尔综合征表型表现的患者,包括巨大血小板以及缺乏瑞斯托霉素诱导的血管性血友病因子结合能力。使用两种与糖蛋白(GP)Ibα链氨基末端结构域中不同表位反应的单克隆抗体,来估计血小板膜上GP Ib分子的数量。在该患者中,一种抗体(LJ-Ib10)的结合量约为正常水平的50%,而另一种抗体(LJ-Ib1)则未检测到结合。先证者的母亲和一个姐妹中,两种抗体的结合量均降至正常水平的约50%,并且她们的血小板表现出约70%的正常血管性血友病因子结合能力。免疫印迹研究证实患者血小板中存在GP Ibα以及GP IX。抗体LJ-Ib10能够从表面标记的蛋白质中免疫沉淀患者的GP Ibα,而LJ-Ib1则不能。因此,先证者的血小板含有结构和功能改变的GP Ib-IX复合物,该复合物缺乏特定抗体表位以及结合血管性血友病因子的能力。相比之下,人α-凝血酶与患者血小板的结合正常,并且可以检测到三类具有高、中、低亲和力的结合位点。这些研究定义了一种独特的伯纳德-索利尔综合征变异形式,并基于天然存在的突变分子提供了证据,表明GP Ibα的氨基末端区域包含一个与高亲和力凝血酶结合位点不同的血管性血友病因子结合结构域。使用具有不同表位特异性的不同单克隆抗体似乎对于正确识别变异型伯纳德-索利尔综合征至关重要。