Harrison Paul M
Dept. of Biology, McGill University, Stewart Biology Building, 1205 Dr, Penfield Ave, Montreal, QC, H3A 1B1, Canada.
BMC Bioinformatics. 2006 Oct 10;7:441. doi: 10.1186/1471-2105-7-441.
Compositionally biased (CB) regions are stretches in protein sequences made from mainly a distinct subset of amino acid residues; such regions are frequently associated with a structural role in the cell, or with protein disorder.
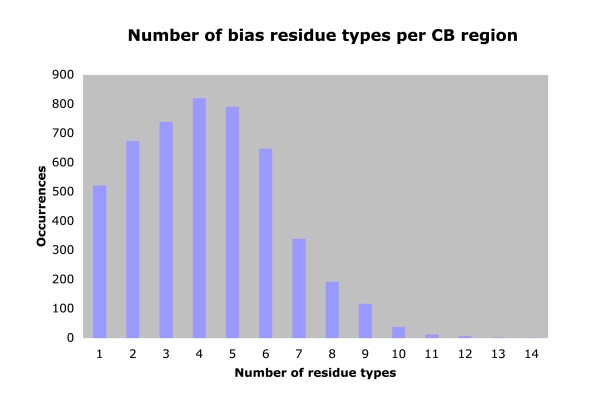
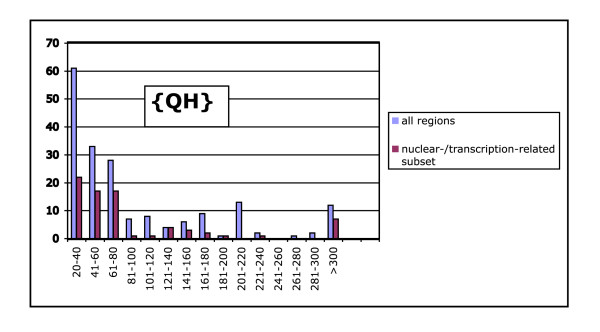
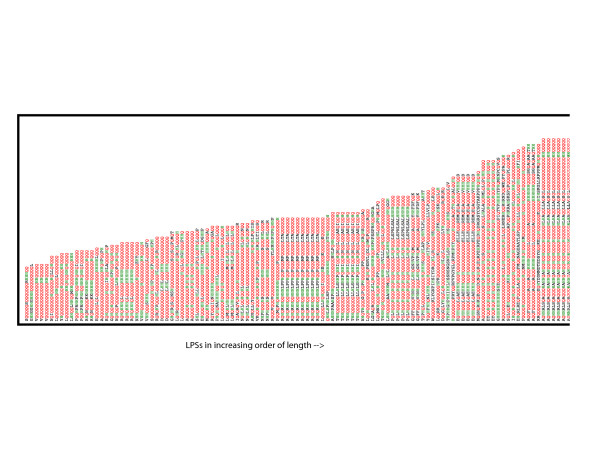
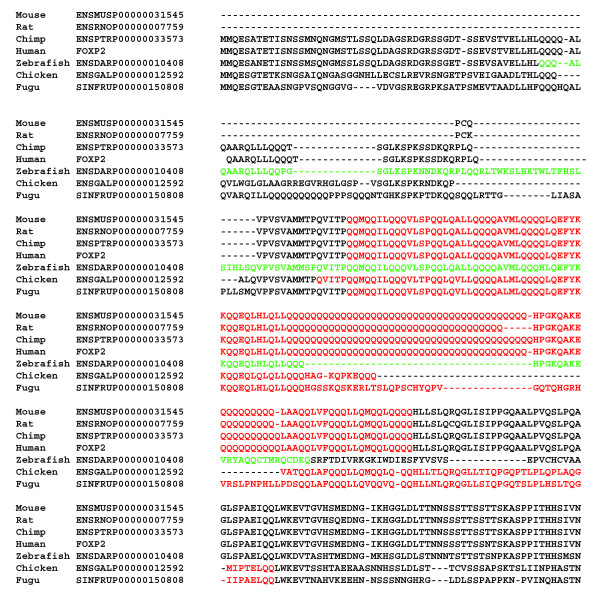
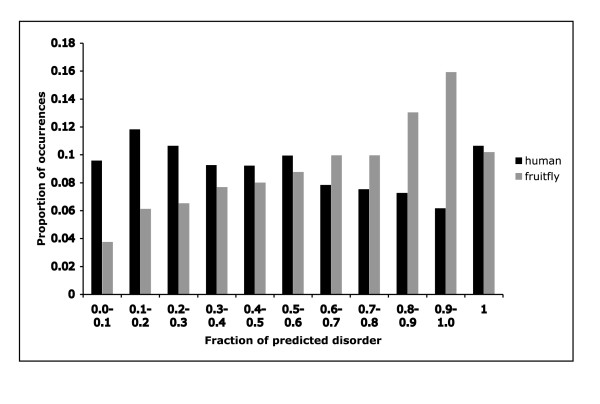
We derived a procedure for the exhaustive assignment and classification of CB regions, and have applied it to thirteen metazoan proteomes. Sequences are initially scanned for the lowest-probability subsequences (LPSs) for single amino-acid types; subsequently, an exhaustive search for lowest probability subsequences (LPSs) for multiple residue types is performed iteratively until convergence, to define CB region boundaries. We analysed > 40,000 CB regions with > 20 million residues; strikingly, nine single-/double- residue biases are universally abundant, and are consistently highly ranked across both vertebrates and invertebrates. To home in subpopulations of CB regions of interest in human and D. melanogaster, we analysed CB region lengths, conservation, inferred functional categories and predicted protein disorder, and filtered for coiled coils and protein structures. In particular, we found that some of the universally abundant CB regions have significant associations to transcription and nuclear localization in Human and Drosophila, and are also predicted to be moderately or highly disordered. Focussing on Q-based biased regions, we found that these regions are typically only well conserved within mammals (appearing in 60-80% of orthologs), with shorter human transcription-related CB regions being unconserved outside of mammals; they are also preferentially linked to protein domains such as the homeodomain and glucocorticoid-receptor DNA-binding domain. In general, only approximately 40-50% of residues in these human and Drosophila CB regions have predicted protein disorder.
This data is of use for the further functional characterization of genes, and for structural genomics initiatives.
成分偏向(CB)区域是蛋白质序列中主要由氨基酸残基的一个独特子集构成的片段;此类区域通常与细胞中的结构作用或蛋白质无序状态相关。
我们推导了一种用于CB区域详尽分配和分类的程序,并将其应用于13个后生动物蛋白质组。首先扫描序列以寻找单个氨基酸类型的最低概率子序列(LPS);随后,对多种残基类型的最低概率子序列(LPS)进行迭代的详尽搜索,直至收敛,以定义CB区域边界。我们分析了超过40,000个CB区域,其中包含超过2000万个残基;引人注目的是,9种单/双残基偏向普遍大量存在,并且在脊椎动物和无脊椎动物中始终排名靠前。为了深入研究人类和黑腹果蝇中感兴趣的CB区域亚群,我们分析了CB区域长度、保守性、推断的功能类别和预测的蛋白质无序状态,并筛选了卷曲螺旋和蛋白质结构。特别是,我们发现一些普遍大量存在的CB区域与人类和果蝇中的转录和核定位有显著关联,并且还被预测为中度或高度无序。聚焦于基于Q的偏向区域,我们发现这些区域通常仅在哺乳动物中具有良好的保守性(出现在60 - 80%的直系同源物中),人类中较短的与转录相关的CB区域在哺乳动物之外不保守;它们还优先与诸如同源结构域和糖皮质激素受体DNA结合结构域等蛋白质结构域相关联。一般来说,这些人类和果蝇CB区域中只有大约40 - 50%的残基具有预测的蛋白质无序状态。
这些数据可用于基因的进一步功能表征以及结构基因组学计划。