Matukumalli Lakshmi K, Grefenstette John J, Hyten David L, Choi Ik-Young, Cregan Perry B, Van Tassell Curtis P
US Department of Agriculture, ARS, Beltsville Agricultural Research Center, Bovine Functional Genomics Laboratory, Beltsville, MD 20705, USA.
BMC Bioinformatics. 2006 Oct 23;7:468. doi: 10.1186/1471-2105-7-468.
Single nucleotide polymorphisms (SNPs) as defined here are single base sequence changes or short insertion/deletions between or within individuals of a given species. As a result of their abundance and the availability of high throughput analysis technologies SNP markers have begun to replace other traditional markers such as restriction fragment length polymorphisms (RFLPs), amplified fragment length polymorphisms (AFLPs) and simple sequence repeats (SSRs or microsatellite) markers for fine mapping and association studies in several species. For SNP discovery from chromatogram data, several bioinformatics programs have to be combined to generate an analysis pipeline. Results have to be stored in a relational database to facilitate interrogation through queries or to generate data for further analyses such as determination of linkage disequilibrium and identification of common haplotypes. Although these tasks are routinely performed by several groups, an integrated open source SNP discovery pipeline that can be easily adapted by new groups interested in SNP marker development is currently unavailable.
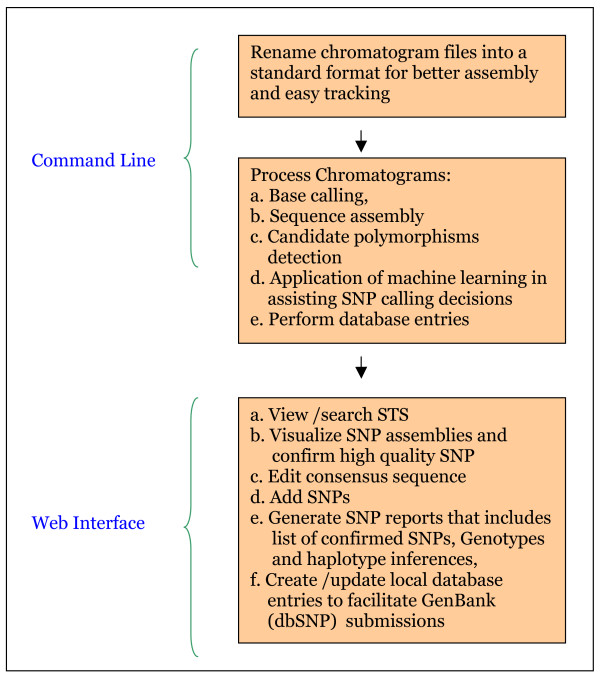
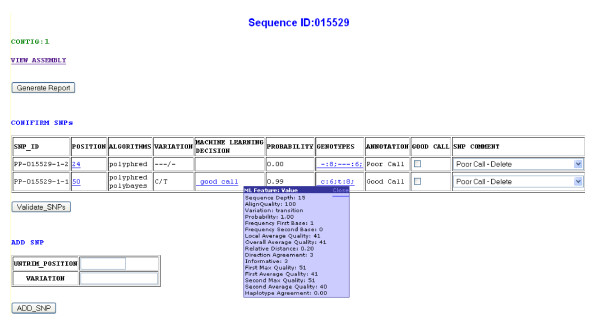
We developed SNP-PHAGE (SNP discovery Pipeline with additional features for identification of common haplotypes within a sequence tagged site (Haplotype Analysis) and GenBank (-dbSNP) submissions. This tool was applied for analyzing sequence traces from diverse soybean genotypes to discover over 10,000 SNPs. This package was developed on UNIX/Linux platform, written in Perl and uses a MySQL database. Scripts to generate a user-friendly web interface are also provided with common queries for preliminary data analysis. A machine learning tool developed by this group for increasing the efficiency of SNP discovery is integrated as a part of this package as an optional feature. The SNP-PHAGE package is being made available open source at http://bfgl.anri.barc.usda.gov/ML/snp-phage/.
SNP-PHAGE provides a bioinformatics solution for high throughput SNP discovery, identification of common haplotypes within an amplicon, and GenBank (dbSNP) submissions. SNP selection and visualization are aided through a user-friendly web interface. This tool is useful for analyzing sequence tagged sites (STSs) of genomic sequences, and this software can serve as a starting point for groups interested in developing SNP markers.
本文所定义的单核苷酸多态性(SNP)是给定物种个体之间或个体内部的单碱基序列变化或短插入/缺失。由于SNP数量丰富且高通量分析技术可用,SNP标记已开始取代其他传统标记,如限制性片段长度多态性(RFLP)、扩增片段长度多态性(AFLP)和简单序列重复(SSR或微卫星)标记,用于多个物种的精细定位和关联研究。为了从色谱图数据中发现SNP,必须组合几个生物信息学程序来生成分析流程。结果必须存储在关系数据库中,以便通过查询进行查询或生成用于进一步分析的数据,如连锁不平衡的测定和常见单倍型的鉴定。尽管这些任务由多个团队常规执行,但目前尚无一个集成的开源SNP发现流程,可供对SNP标记开发感兴趣的新团队轻松采用。
我们开发了SNP - PHAGE(具有用于在序列标签位点内鉴定常见单倍型(单倍型分析)和向GenBank(-dbSNP)提交数据的附加功能的SNP发现流程)。该工具用于分析来自不同大豆基因型的序列痕迹,以发现超过10,000个SNP。此软件包是在UNIX/Linux平台上开发的,用Perl编写,并使用MySQL数据库。还提供了用于生成用户友好型网页界面的脚本以及用于初步数据分析的常见查询。该团队开发的用于提高SNP发现效率的机器学习工具作为此软件包的一个可选功能集成其中。SNP - PHAGE软件包可在http://bfgl.anri.barc.usda.gov/ML/snp - phage/上以开源形式获取。
SNP - PHAGE为高通量SNP发现、扩增子内常见单倍型的鉴定以及向GenBank(dbSNP)提交数据提供了一种生物信息学解决方案。通过用户友好型网页界面辅助进行SNP选择和可视化。该工具对于分析基因组序列的序列标签位点(STS)很有用,并且此软件可作为对开发SNP标记感兴趣的团队的起点。